TẦM QUAN TRỌNG CỦA THEO DÕI HUYẾT ĐỘNG ĐỂ CHẨN ĐOÁN VÀ ĐỊNH NGHĨA ARDS.
Hội chứng suy hô hấp cấp (ARDS) là tình trạng viêm với tổn thương lan tỏa hàng rào phế nang-mao mạch dẫn đến sung huyết mao mạch, xuất huyết trong phế nang, hình thành màng hyalin và phù nề mô kẽ phế nang [1]. ARDS là một dạng phù phổi không do tim, vì tích tụ dịch không phải từ việc tăng áp lực thủy tĩnh gây hiện tượng siêu lọc mao mạch do tăng áp lực nhĩ trái, mà là hậu quả của sự suy giảm tính thấm của hàng rào phế nang-mao mạch. Loại trừ nguồn gốc suy hô hấp do tim là một tiêu chuẩn chẩn đoán cần thiết cho ARDS. Trong định nghĩa của Hội nghị Đồng thuận Mỹ-Âu (AECC) trước đây về ARDS (được thông qua năm 1994), giá trị của áp lực động mạch phổi bít (Ppao) thấp hơn 18 mmHg là bắt buộc để xác định chẩn đoán [2]. Tuy nhiên, vì giá trị Ppao cao do suy tim hoặc quá tải dịch truyền có thể cùng tồn tại với ARDS và do việc sử dụng catheter động mạch phổi (PAC) liên tục giảm, nên yêu cầu về giá trị Ppao đã được loại bỏ khỏi “định nghĩa Berlin” hiện đang được áp dụng của ARDS [3]. Tuy nhiên, định nghĩa Berlin nói rằng “suy hô hấp không được giải thích đầy đủ do suy tim hoặc quá tải dịch bằng tất cả các dữ liệu có sẵn” và “nếu không có yếu tố nguy cơ ARDS rõ ràng, cần phải có một số đánh giá khách quan để loại trừ khả năng phù thủy tĩnh. ” Kết luận, theo dõi huyết động chức năng là cần thiết để loại trừ nguồn gốc tim của phù phổi và để xác định chẩn đoán hội chứng.
TẦN SUẤT VÀ NGUYÊN NHÂN CỦA BẤT ỔN HUYẾT ĐỘNG TRONG ARDS.
Phần lớn bệnh nhân ARDS phát triển huyết động không ổn định [4], và hơn một nửa trong số họ cần sử dụng thuốc vận mạch [5]. Suy tuần hoàn và cần dùng thuốc vận mạch có liên quan nhiều hơn đến nguy cơ tử vong hơn là mức độ nghiêm trọng của tình trạng giảm oxy máu [6]. Một số yếu tố góp phần vào sự phát triển của huyết động không ổn định ở bệnh nhân ARDS:
Tác dụng độc tính trực tiếp của giảm oxy máu và nhiễm toan lên chức năng cơ tim. Oxy, ngoài chức năng thiết yếu của nó trong chuyển hóa năng lượng tim, còn tham gia vào các quá trình khác ảnh hưởng đến chức năng tế bào cơ tim, chẳng hạn như điều hòa biểu hiện gen tim và tạo ra các loại oxy phản ứng, có thể gây ra tổn thương tế bào không thể phục hồi [7]. Mặt khác, nhiễm toan nội bào có liên quan đến suy giảm sức co bóp của tim và thậm chí có thể gây hoại tử cơ tim [8].
Tổn thương tim mạch liên quan đến nhiễm khuẩn huyết: nhiễm trùng huyết là nguyên nhân thường gặp nhất của ARDS [9]. Cơ chế của suy tim mạch trong nhiễm trùng huyết là sốc phân bố, thay đổi tính thấm thành mạch, tăng nhu cầu oxy của mô và rối loạn chức năng cơ tim.
Các bệnh tim mạch mắc đồng thời có từ trước.
Rối loạn chức năng mạch phổi và rối loạn chức năng thất phải (RV).
Tác dụng huyết động của thông khí áp lực dương.
Hai điểm sau, về sự liên quan lâm sàng và sinh lý bệnh của chúng, sẽ được thảo luận trong các đoạn dành riêng.
Rối loạn chức năng mạch máu phổi.
ARDS được đặc trưng bởi tổn thương tuần hoàn phổi dẫn đến phá hủy mao mạch phổi, tăng sức cản mạch phổi (PVR) và tăng áp động mạch phổi (PH), như được Zapol mô tả lần đầu tiên vào năm 1977 [10, 11]. Tăng áp phổi (PH) được xác định bằng áp lực động mạch phổi trung bình (mPAP) > 25 mmHg [12]. Giá trị tiên lượng của PH vẫn chưa rõ ràng: một số nghiên cứu tìm thấy mối liên hệ đáng kể của giá trị mPAP với tỷ lệ tử vong [13], trong khi những nghiên cứu khác thì không [14– 16]. Thuật ngữ rối loạn chức năng mạch phổi (PVD – pulmonary vascular dysfunction) đã được đề xuất để chỉ sự tăng PVR được định nghĩa là tăng gradient xuyên phổi (mPAP-Ppao) [17] và/ hoặc chỉ số sức cản mạch phổi (mPAP-Ppao /chỉ số tim) [18]. Một lần nữa, dữ liệu về mối liên quan giữa tăng PVR và tỷ lệ tử vong là mâu thuẫn nhau [19]. Bull và cộng sự đã thực hiện một phân tích hậu kiểm của Thử nghiệm điều trị dịch truyền và catheter (FACTT) và phát hiện ra rằng, trong phân nhóm 501 bệnh nhân được quản lý bằng PAC, PVD là một yếu tố dự đoán độc lập về kết quả bất lợi. Tuần hoàn phổi được đặc trưng bởi compliance cao và sức cản dòng máu thấp, và vì lý do này, mối quan hệ giữa áp lực đẩy và lưu lượng là đường cong tuyến tính, trong khi trong tuần hoàn hệ thống, mối quan hệ này là tuyến tính.
Điều này có nghĩa là trong điều kiện bình thường, PAP tăng dẫn đến lưu lượng máu phổi tăng không cân xứng không chỉ do tăng áp lực đẩy (tức là sự chênh lệch giữa mPAP và áp lực tâm nhĩ trái) mà còn do sự căng phồng của các mạch và sự huy động các mao mạch đã đóng trước đó. Ngược lại, giảm cung lượng tim sẽ liên quan đến giá trị PVR được tính toán cao hơn, ngay cả khi trương lực vận mạch không thay đổi (Hình 34.1). Trong ARDS, mọi thứ thậm chí còn trở nên phức tạp hơn, vì mối quan hệ giữa lưu lượng máu phổi và PAP thay đổi do sự hiện diện của PVD: do đó, những thay đổi trong giá trị PVR được tính toán có thể đặc biệt khó giải thích [20, 21].
Ở bệnh nhân ARDS, các cơ chế sau đây góp phần vào sự phát triển của PVD:
Chứng co mạch phổi do giảm oxi máu (HPV). Sự cải thiện oxy hóa dẫn đến giảm HPV và do đó làm giảm PVR [22, 23]. Carbon dioxide cũng gây ra tác dụng co mạch đáng kể trên mạch phổi [24]: tăng CO2 máu có liên quan đến tăng PVR và do đó nguy cơ rối loạn chức năng thất phải cao hơn [25, 26]. Sự co mạch cũng được thúc đẩy bởi tổn thương nội mô, gây ra sự mất cân bằng giữa chất trung gian giãn mạch và chất co mạch mà nghiêng về cho tình trạng sau hơn [27]. Do đó, nó được quan sát thấy giảm khả năng thích ứng với những thay đổi trong lưu lượng máu [4].
Dự trữ mao mạch cạn kiệt, do huyết khối hoặc chèn ép bởi phù mô kẽ của động mạch và mao mạch phổi [28]. Tổn thương nội mô trong tổn thương phổi cấp tính gây kích hoạt cục bộ đông máu [27] và tiêu sợi huyết đóng vai trò tiên lượng và di truyền bệnh đáng kể trong ARDS [29].
Tăng sinh sợi và tái tạo mạch máu (giai đoạn cuối của ARDS).
Ảnh hưởng của thông khí áp lực dương: sự thay đổi áp suất đường thở ảnh hưởng mạnh đến lưu lượng máu đến phổi bằng cách ảnh hưởng đến áp lực mạch xuyên thành mạch và sự phân bố lưu lượng máu qua các vùng phổi khác nhau [30]. Việc áp dụng áp lực dương cuối kỳ thở ra (PEEP) và hậu quả là tăng áp lực đường thở trung bình (Paw) có ảnh hưởng sâu sắc đến PAP, qua trung gian của những thay đổi về áp lực xuyên phổi và thể tích phổi. PVR có mối quan hệ hình chữ U với thể tích phổi, tăng cả ở thể tích phổi thấp do chèn ép các mạch ngoài phế nang và ở thể tích phổi rất cao do giãn các mạch phế nang [31]. Do đó, tác động của PEEP lên PVR phụ thuộc vào sự cân bằng giữa việc huy động các khu vực bị xẹp và tăng bơm của các đơn vị phổi đã mở. Nếu việc tuyển dụng chiếm ưu thế, sự căng phồng của các mạch ngoài phế nang và sự giảm đồng thời của HPV sẽ dẫn đến giảm PVR. Nếu tình trạng tăng bơm diễn ra, sự chèn ép của các mao mạch trong phế nang tạo ra các điều kiện của vùng West 1 và 2, do đó làm tăng PVR [30, 32] và hậu tải RV [33]. Cuối cùng, vùng West 2 gây ra sự chuyển hướng của dòng máu về các đơn vị phế nang thông khí kém, làm quá trình trao đổi khí trở nên tồi tệ hơn.
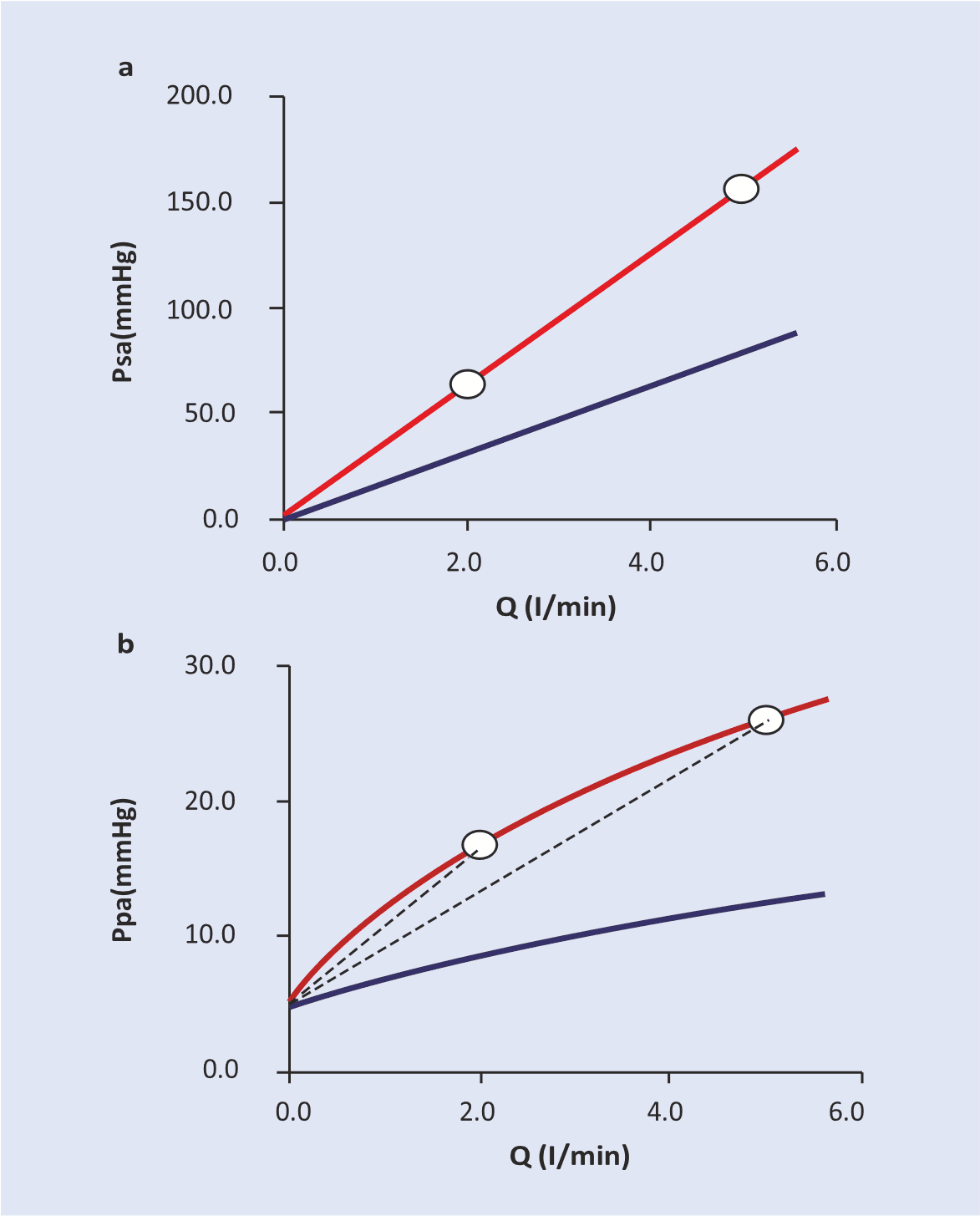
Hình 34.1. Áp lực động mạch trung bình được biểu đồ hóa dựa trên lưu lượng (cung lượng tim) trong tuần hoàn hệ thống (a) và phổi (b). Đường cong màu xanh lam trong mỗi bảng biểu thị tình trạng bình thường của hệ tuần hoàn và đường cong màu đỏ biểu thị tình trạng tăng áp. (a) Trong tuần hoàn hệ thống, đồ thị áp suất trung bình (P) -lưu lượng (Q) được mô tả tốt như một mối quan hệ tuyến tính (như định luật Ohmic). Hai điểm được xác định (vòng tròn mở) cho thấy lần lượt cung lượng tim bình thường và cung lượng tim giảm, trong tình trạng tăng áp. Ở mỗi cung lượng tim này, rõ ràng là tỷ lệ P so với Q là như nhau và do đó có thể được sử dụng để dễ dàng mô tả sức cản của tuần hoàn hệ thống. (b) Trong tuần hoàn phổi, đồ thị của áp suất trung bình so với dòng chảy là một đường cong với điểm chặn trên trục áp suất bằng với áp suất tâm nhĩ trái. Đường cong màu xanh biểu thị đường cong lưu lượng áp suất bình thường (phổi khỏe mạnh), trong khi đường cong màu đỏ biểu thị đường cong lưu lượng áp suất khi có tăng áp phổi do thiếu oxy. Hai điểm được xác định (vòng tròn mở) cho thấy cung lượng tim bình thường và cung lượng tim giảm tương ứng, trong tình trạng tăng huyết áp. Tại mỗi cung lượng tim, sức cản mạch máu phổi, Ppa-LAP/ Q, được minh họa dưới dạng độ dốc của đường chấm. Mặc dù hai điểm nằm trên cùng một đường cong lưu lượng áp lực, nhưng sức cản mạch phổi được tính toán là khác nhau ở các cung lượng tim khác nhau. Psa, áp lực động mạch hệ thống (trung bình); Ppa, áp lực động mạch phổi (trung bình); Q, cung lượng tim (lưu lượng).
Suy giảm chức năng thất phải.
Các tác động tổng hợp của ức chế cơ tim do nhiễm trùng huyết, tác động huyết động của thông khí áp lực dương, tác động có hại của giảm oxy máu đối với sức co bóp cơ tim và PVD có thể ảnh hưởng nghiêm trọng đến chức năng tim, dẫn đến rối loạn chức năng thất phải (RVD) và tâm phế cấp (ACP). RV rất nhạy cảm với những thay đổi của hậu tải: compliance RV và có thành mỏng phù hợp hơn để tránh được sự gia tăng đáng kể của tiền tải nhưng kém chịu đựng sự gia tăng cấp tính của hậu tải [34, 35]. Trong ARDS, sự gia tăng hậu tải RV dẫn đến sự không phù hợp giữa tăng nhu cầu oxy và giảm áp lực tưới máu động mạch vành phải [36]. Suy giảm tống máu RV làm giảm sự hồi lưu của tĩnh mạch phổi về LV, tăng thể tích RV cuối tâm thu và cuối tâm trương, và giảm compliance tâm trương LV, thông qua sự phụ thuộc lẫn nhau của thất. Hậu quả của sự giảm phân suất tống máu LV dẫn đến giảm lưu lượng máu vành RV với hậu quả là thiếu máu cục bộ RV: vòng luẩn quẩn này duy trì và khuếch đại hơn nữa rối loạn chức năng RV, cuối cùng dẫn đến ACP, bao gồm sự tách rời đáng kể động mạch thất giữa RV và tuần hoàn phổi [24]. Kết quả rõ ràng là giảm cung lượng tim và khả năng đáp ứng thể tích kém, có nguy cơ quá tải dịch.
Dựa trên các tiêu chí siêu âm tim, có thể xác định ba bước tuần tự của suy giảm chức năng RV ở bệnh nhân ARDS:
RV giãn: RV/ LV vùng cuối tâm trương > 0,6.
Rối loạn chức năng RV: TAPSE <16 mm.
Tâm phế cấp (ACP): sự liên quan của giãn RV với chuyển động nghịch lý của vách liên thất ở cuối tâm thu, cái gọi là “hình chữ D” của LV [37].
Ảnh hưởng huyết động của thông khí áp lực dương.
Phổi và tim nằm trong khoang ngực và được bao quanh bởi áp lực trong lồng ngực (ITP) (tương ứng là màng phổi và màng tim). Hiệu ứng huyết động của thông khí áp lực dương có thể được chia thành các hiệu ứng “trạng thái ổn định” và “pha”.

Hình 34.2 Hiệu ứng giả định của áp suất màng phổi trong quá trình thở có kiểm soát và tự phát đối với áp lực xuyên mạch máu. Trong hình minh họa này, phổi căng ra đến cùng một áp lực xuyên phổi 30 cmH2O bởi ba sự kết hợp của áp lực phế nang và màng phổi. Nếu áp suất lòng vi mạch tương đương với 15 cmH2O và áp lực kẽ được giả định bằng áp suất màng phổi, thì áp suất xuyên mạch máu lọc chất lỏng (PTV) trong ví dụ cực đoan này về mặt lý thuyết sẽ nằm trong khoảng từ 5 đến 25 cmH2O
Hiệu ứng trạng thái ổn định phụ thuộc cả vào các yếu tố cơ học và thần kinh thể dịch, phản ánh tác động của sự thay đổi bền vững các điều kiện hô hấp [38].
Chúng phụ thuộc vào sự gia tăng của Paw trung bình, được xác định bởi mức PEEP (quan trọng nhất), thời gian chu kỳ làm việc và áp lực đẩy của [39]. Việc áp dụng PEEP có liên quan đến việc giảm cung lượng tim, biểu hiện rõ ràng hơn khi có giảm thể tích tuần hoàn. Lời giải thích phổ biến là sự gia tăng ITP chuyển thành sự gia tăng áp lực tâm nhĩ phải (Pra), gây ra giảm chênh áp cho sự hồi lưu của tĩnh mạch, tức là sự khác biệt giữa áp lực đổ đầy hệ thống trung bình (Pmsf) và Pra [40]. Tuy nhiên, cách giải thích này có vẻ quá đơn giản. Thật vậy, người ta đã chứng minh rằng PEEP không chỉ làm tăng Pra mà còn làm tăng Pmsf thông qua việc tăng cường phản xạ của trương lực tĩnh mạch (nguyên nhân chuyển máu từ thể tích không bị stress sang thể tích stress) [41] và thông qua việc tăng áp lực ổ bụng (với sự đè ép của hồ chứa tĩnh mạch tạng) [42]. Điều này cho thấy rằng những thay đổi trong cung lượng tim có thể xảy ra độc lập với những thay đổi của độ chênh áp suất cho sự hồi lưu của tĩnh mạch [43]. Ngoài ra, Marini et al. đã chứng minh rằng PEEP có ít ảnh hưởng trực tiếp đến chức năng tâm thất [44].
Tác dụng thoáng qua phụ thuộc vào những thay đổi có chu kỳ về thể tích phổi / thành ngực và áp lực đường thở / lồng ngực trong chu kỳ hô hấp, có dấu hiệu ngược lại trong quá trình thông khí tự phát và có kiểm soát. Cả trong quá trình thở vào áp suất âm và dương, sự giãn nở của phổi sẽ trực tiếp nén tim trong hố tim, làm ảnh hưởng đến quá trình tiền tải. Trong quá trình thở tự nhiên, cho dù ở bệnh nhân thở máy hay không thở máy, sự giảm ITP sẽ làm tăng gradient hồi lưu của tĩnh mạch (Pmsf – Pra), gây ra tăng tiền tải RV và giãn RV. Loại thứ hai (giãn RV) có liên quan đến giảm sự comliance và đổ đầy của LV, dẫn đến giảm thể tích nhát bóp LV trong quá trình thở vào, giải thích cho sự giảm áp lực động mạch tâm thu sinh lý khi hít vào [45, 46]. Ngược lại, thông khí áp lực dương làm tăng ITP và thể tích phổi theo chu kỳ, cả hai đều làm giảm hồi lưu của tĩnh mạch. Sự gia tăng áp lực trong phổi gây ra tăng PVR và sau đó là hậu tải RV, với giảm thể tích nhát bóp RV. Những thay đổi ngược lại sẽ xảy ra trên buồng tim trái: tiền tải LV có xu hướng tăng vì máu bị “ép ra” khỏi mạch phổi, trong khi hậu tải LV cải thiện do giảm áp lực xuyên thành động mạch chủ. Do đó, thể tích nhát bóp LV sẽ tăng trong quá trình bơm vào với áp suất dương và giảm khi thở ra. Sự thay đổi áp lực màng phổi đối với một thay đổi nhất định trong Paw (tức là phần tăng Paw “truyền” đến ITP) phụ thuộc vào thể tích thông khí và vào compliance của thành ngực (CCW) so với compliance của phổi (CL) [47] theo công thức: ΔPpl/ ΔPaw = CL/ (CL + CCW) [48]. Ở các đối tượng bình thường, vì CL và CCW tương tự nhau, ITP nên tăng khoảng 50% mức tăng của Paw trung bình [47, 48]. Sự gia tăng Paw tương tự sẽ gây ra những tác động hoàn toàn khác nhau ở một bệnh nhân có phổi rất cứng và thành ngực bình thường (ví dụ, với ARDS nguyên phát) so với bệnh nhân có phổi compliance bình thường và thành ngực cứng (ví dụ, với khí phế thũng hoặc tăng áp lực trong ổ bụng). Trong trường hợp đầu tiên, sự thay đổi trong ITP gây ra bởi sự gia tăng nhất định trong Paw sẽ ở mức khiêm tốn và sẽ có tác động huyết động hạn chế; trong trường hợp thứ hai, sự gia tăng Paw sẽ dẫn đến sự gia tăng ITP lớn hơn nhiều với các tác động huyết động rõ rệt hơn.
Ảnh hưởng của các nhịp thở tự nhiên.
Những bệnh nhân thở tự do với nhịp hô hấp cao có thể tạo ra sự thay đổi áp lực màng phổi âm lớn trong khi cố gắng thở, dẫn đến thay đổi áp suất xuyên phổi cao hơn và tăng độ căng và độ dãn của phổi [49]. Hơn nữa, cùng một sự thay đổi áp lực xuyên phổi có thể liên quan đến các giá trị tuyệt đối hoàn toàn khác nhau của áp lực phế nang (Palv) [50]. Thực tế, khi có nỗ lực thở tự phát, Palv có thể giảm xuống thấp hơn giá trị áp lực cuối kỳ thở ra và thậm chí có thể trở nên âm trong toàn bộ chu kỳ thở, chuyển thành tăng áp lực xuyên mạch phổi (tức là chênh lệch giữa áp lực mao mạch nội mạch và Palv) [ 51] (Hình 34.2). Điều này, với sự gia tăng tính thấm thành mạch, gây ra rò rỉ mạch máu và phù nề mô kẽ, góp phần làm nặng hơn và duy trì tổn thương phổi [49].
CÁC NGUYÊN LÝ QUẢN LÝ HUYẾT ĐỘNG.
Quản lý huyết động ở bệnh nhân ARDS nên được hướng dẫn bởi ba nguyên tắc cơ bản, đều quan trọng như nhau nhưng đôi khi khó đạt được cùng một lúc:
Tối ưu hóa việc cung cấp oxy
Tránh quá tải DỊCH
Phòng ngừa suy giảm chức năng RV.
Tối ưu hóa phân phối oxy.
Mục tiêu cuối cùng của hỗ trợ tim phổi là tối ưu hóa quá trình tưới máu và phân phối oxy đến các mô ngoại vi, nhằm đáp ứng nhu cầu trao đổi chất của cơ thể. Cung cấp oxy (DO2) là thể tích oxy được cung cấp đến giường mạch hệ thống trong một phút và là sản phẩm của hàm lượng oxy trong động mạch (CaO2) và cung lượng tim (CO): DO2 = CaO2 × CO. CaO2 được định nghĩa là tổng lượng oxy được vận chuyển bởi hemoglobin và oxy hòa tan trong huyết tương, theo công thức CaO2 = (Hb × 1,39 × SaO2) + (P aO2 × 0,003), trong đó Hb là mức hemoglobin, SaO2 là độ bão hòa của động mạch, PaO2 là sức căng riêng phần oxy của động mạch, và 0,003 là hệ số hòa tan của oxy trong huyết tương người. Vì đường cong phân ly oxyhemoglobin trở nên tương đối bằng phẳng khi SaO2 > 90%, sự gia tăng thêm của PaO2 có tác động tương đối ít đến SaO2 hoặc CaO2 trong khoảng này. Ngoài ra, lượng oxy hòa tan không đáng kể, một khi máu động mạch đã bão hòa hoàn toàn, cách duy nhất để tăng CaO2 là tăng nồng độ hemoglobin. Do đó, nhắm vào nồng độ hemoglobin cao hơn ở những bệnh nhân ARDS giảm oxy máu nghiêm trọng sẽ cải thiện CaO2, làm tăng khả năng vận chuyển oxy của máu động mạch (Russell 1999). Các hướng dẫn của Tổ chức Hỗ trợ Sự sống Ngoài cơ thể khuyến nghị duy trì mức Hb từ 12–14 gr/ dL và hematocrit bình thường ở những bệnh nhân đang điều trị ECMO do suy hô hấp giảm oxy máu nặng [52]. Tuy nhiên, một số tác dụng phụ của việc truyền hồng cầu đã được báo cáo ở những bệnh nhân bị tổn thương phổi cấp tính [53–55], và ngưỡng Hb tối ưu cho việc truyền máu vẫn còn là một vấn đề còn bỏ ngỏ. Thành phần khác của DO2 là CO, là sản phẩm của thể tích đột quỵ (SV) và nhịp tim (HR): CO = SV × HR. CO có thể được đo bằng nhiều thiết bị xâm lấn bao gồm PAC và hệ thống pha loãng nhiệt qua phổi (ví dụ, PiCCO hoặc Vigileo) và các thiết bị phân tích dạng sóng áp lực động mạch (ví dụ, LiDCO, PiCCO, FloTrac, MostCare, v.v.). Trong số các kỹ thuật không xâm lấn hiện có để ước tính CO, siêu âm tim là phổ biến nhất [56]: sự thống nhất rất tốt giữa CO được ước tính bằng siêu âm tim qua lồng ngực và được đo bằng phương pháp nhiệt độ qua phổi đã được báo cáo ở những bệnh nhân nặng, thở máy [57]. Tuy nhiên, những bệnh nhân có mức PEEP cao có thể có “cửa sổ âm thanh” kém và độ tin cậy và khả năng tái tạo của các phát hiện siêu âm tim phụ thuộc nhiều vào chuyên môn của người thực hiện. Ở những bệnh nhân CO không đủ, cần đánh giá từng yếu tố quyết định sinh lý của nó (tải trước, hậu tải và co bóp). Đặc biệt, một bước quan trọng là phải hiểu liệu bệnh nhân có cần truyền dịch để tối ưu hóa tiền tải hay không.
Một số phương pháp có sẵn để đánh giá khả năng đáp ứng dịch truyền (FR):
Thách thức dịch truyền: đánh giá đầy đủ FR đòi hỏi phải tích hợp các biến đổi của áp lực đổ đầy tim (Pra và/ hoặc Ppao) với các biến thiên CO sau thử thách dịch truyền (ví dụ, truyền 250 mL hoặc 3 mL/ Kg tinh thể trong 5 phút) [ 58, 59]. Pra (hay còn gọi là áp suất tĩnh mạch trung tâm hoặc CVP) và chỉ Ppao đơn độc không phải là chỉ số chính xác của tiền tải và FR [60–62], vì giá trị tuyệt đối của chúng (được đo so với áp suất khí quyển) không nhất thiết phản ánh áp suất xuyên thành, đó là chênh lệch áp suất giữa bên trong và bên ngoài mạch máu (trong trường hợp này là ITP) và là áp suất liên quan đối với FR. Sự phân biệt quan trọng này rất thường bị bỏ qua. Chính áp suất trung tâm so với khí quyển chứ không phải áp suất xuyên thành xác định áp suất trở về cho sự hồi lưu của tĩnh mạch và do đó là áp suất siêu lọc mao mạch thủy tĩnh trong các mô ngược dòng [63]. Tuy nhiên, Pra hoặc Ppao tuyệt đối cao thực sự có thể tương ứng với các giá trị áp suất xuyên thành thấp, nếu ITP tăng lên đáng kể. Nói cách khác, một bệnh nhân có Pra cao có thể bị phù ngoại vi và sung huyết gan nhưng vẫn có sự gia tăng đáng kể CO sau khi truyền dịch [64]. Một “phiên bản thay thế” của test thách thức dịch truyền là test nâng cao chân thụ động (PLR): nâng chân gây ra sự chuyển dịch của máu tĩnh mạch từ vùng cơ thể thấp hơn đến khoang ngực, hoạt động như một thử thách thể tích tự bản thân có thể đảo ngược. Sự gia tăng ít nhất 10% lưu lượng máu động mạch chủ hoặc chỉ số tim là giá trị ngưỡng tốt nhất để phát hiện FR sau PLR [65].
Các chỉ số động: như đã mô tả trước đây, các biến đổi theo chu kỳ của ITP trong quá trình thông khí áp lực dương gây ra các biến thiên đồng thời của thể tích nhát bóp LV, tăng trong khi hít vào và giảm khi thở ra [66]. Tình trạng thể tích của bệnh nhân ảnh hưởng sâu sắc đến mức độ của những thay đổi này, biểu hiện rõ rệt hơn khi có giảm thể tích tuần hoàn. Cả sự biến thiên áp suất xung (PPV) và sự biến thiên thể tích nhát bóp (SVV) đều được sử dụng để đánh giá FR. Áp lực xung (PP) là hiệu số giữa huyết áp tâm thu và huyết áp tâm trương, và PPV được tính bằng cách chia PP lớn nhất cho PP trung bình: giá trị > 13% là một dự báo tốt của FR [67]. SVV được đo bằng sóng động mạch xâm lấn tối thiểu từ phân tích (ví dụ, LiDCO, PiCCO, FloTrac, MostCare, v.v.) hoặc Doppler thực quản và được tính bằng cách chia sự khác biệt giữa SV tối đa và SV tối thiểu cho mức trung bình của chúng trong khoảng thời gian 30s . Tuy nhiên, một số hạn chế của PPV và SVV đặc biệt có liên quan ở bệnh nhân ARDS:
Chúng yêu cầu không có nỗ lực thở tự phát.
Thể tích thông khí nhỏ (<8 mL/ Kg) và/ hoặc mức độ tuân thủ của phổi thấp có thể không tạo ra những thay đổi đầy đủ trong ITP [68, 69]. Nhưng nếu PPV hoặc SVV > 13% với nhịp thở thể tích thông khí thấp, chúng vẫn dự đoán FR.
PPV dự đoán FR kém trong trường hợp PH vì ACP gây ra PPV của chính nó.
Chúng có thể kém tin cậy hơn khi có rối loạn chức năng RV đáng kể vì lý do tương tự như PH.
Test tắc nghẽn cuối kỳ thở ra (EEO): nó bao gồm thực hiện tạm dừng thở ra trong 15s, do đó ngăn cản trở lực tuần hoàn đối với việc làm đầy RV và LV và hoạt động như một loại thử thách dịch truyền; test độc lập với sự tuân thủ của hệ thống hô hấp. Sự gia tăng CO do EEO gây ra lên 5% dường như là một yếu tố dự báo tốt về FR ở bệnh nhân ARDS [70].
Khi tình trạng thể tích của bệnh nhân đủ, có thể cần dùng các thuốc co bóp để cải thiện sức co bóp cơ tim và tăng CO. Đánh giá chính xác mức độ đầy đủ của DO2 và tưới máu mô là bắt buộc ở mỗi bệnh nhân nguy kịch: dấu hiệu lâm sàng và test của việc cung cấp oxy không đủ là da nổi bông, giảm thời gian đổ đầy mao mạch, giảm lượng nước tiểu, khử bão hòa oxy hỗn hợp tĩnh mạch và tăng lactate máu. Độ bão hòa oxy trong tĩnh mạch hỗn hợp (SvO2) đặc biệt mang tính thông tin, vì nó phụ thuộc vào tỷ lệ giữa DO2 và lượng oxy tiêu thụ (VO2): SvO2 giảm xuống dưới giá trị bình thường 70% có thể do cung cấp oxy không đủ, tiêu thụ oxy quá mức, hoặc cả hai. Do đó, ở những bệnh nhân có SvO2 giảm, điều quan trọng không chỉ là cải thiện DO2 mà còn giảm tiêu thụ oxy của mô, ví dụ, bằng cách tối ưu hóa an thần và điều trị sốt và đau.
Trong trường hợp có giảm độ bão hòa oxy tĩnh mạch trộn và/ hoặc tăng lactat máu, cần theo dõi huyết động chính xác. Cần nhấn mạnh hai vấn đề:
Đo độ bão hòa oxy tĩnh mạch hỗn hợp “thực” yêu cầu đặt PAC. Nếu không có PAC, độ bão hòa oxy của máu thu thập thông qua catheter tĩnh mạch trung tâm đặt trong tâm nhĩ phải có thể được sử dụng thay thế cho độ bão hòa oxy tĩnh mạch hỗn hợp. Thật vậy, trong khi độ bão hòa oxy tĩnh mạch trung tâm có xu hướng đánh giá quá cao độ bão hòa oxy tĩnh mạch hỗn hợp, thông qua các biến thiên của nó phản ánh một cách đáng tin cậy các biến thiên của máu tĩnh mạch hỗn hợp [71].
Ở những bệnh nhân nhiễm trùng huyết, độ bão hòa oxy trộn tĩnh mạch và tĩnh mạch trung tâm có thể bị nhầm lẫn về bình thường hoặc thậm chí tăng cao. Sự không đồng nhất của tưới máu vi tuần hoàn, sự mở của các shunt vi tuần hoàn và rối loạn chức năng ty thể dẫn đến sự tách biệt bệnh lý giữa hệ thống và vi tuần hoàn [72] với khả năng chiết xuất và sử dụng oxy bị giảm.
Tránh quá tải dịch
Như đã đề cập trước đây, ARDS là một dạng phù phổi không do tim, tính thấm cao, có thể trở nên tồi tệ hơn khi truyền dịch ngoại sinh. Nhiều bằng chứng cho thấy cân bằng dịch dương sau giai đoạn hồi sức ban đầu là một yếu tố tiên lượng bất lợi độc lập ở những bệnh nhân nặng [73–75]. Một chỉ số thú vị về sự tích tụ dịch trong phổi là nước phổi ngoài mạch (EVLW), cụ thể là lượng nước chứa trong phổi bên ngoài hệ mạch phổi. Nó tương ứng với tổng lượng dịch kẽ, nội bào, phế nang và bạch huyết, không bao gồm tràn dịch màng phổi [76]. Giá trị bình thường của EVLW là <10 mL/ Kg [77] và nó được điều hòa một cách chặt chẽ bởi một số cơ chế: hệ thống dẫn lưu bạch huyết giữ cho nền mô khô ráo, trong khi các điểm nối chặt chẽ và hệ thống vận chuyển ion tích cực của tế bào phế nang bảo vệ không gian phế nangn khỏi bị ngập lụt [78]. Định luật Starling chi phối sự thanh thải thực của phế nang và dịch kẽ: tăng áp lực thủy tĩnh mao mạch phổi hoặc giảm áp lực keo làm tăng EVLW. Chỉ số tính thấm thành mạch phổi tăng (PVPI = tỷ số giữa EVLW và thể tích máu phổi) được coi là một dấu hiệu nhận biết của ARDS. Tăng áp động mạch phổi, suy tim và giữ nước do ảnh hưởng nội tiết (tức là hoạt hóa hệ thống angiotensin-aldosterone ở thận) thường cùng tồn tại với ARDS và thúc đẩy phù thủy tĩnh chồng lên dịch tiết ban đầu [79]. Cuối cùng, liệu pháp điều trị dịch truyền (đặc biệt với các dịch tinh thể) cũng có thể góp phần làm cho phổi bị quá tải. Trong một vòng luẩn quẩn, tình trạng quá tải dịch làm giảm áp suất thẩm thấu keo trong mao mạch phổi, làm tình trạng phù phổi trầm trọng hơn. Theo định nghĩa, trong ARDS phù phổi không phải do suy tim hoặc quá tải dịch, nhưng vì những lý do nêu trên, tăng tiền tải thất trái không loại trừ ARDS. Thật vậy, các nghiên cứu trên mô hình động vật về tổn thương phổi cấp chỉ ra rằng phù phổi giảm nếu hạ áp lực nhĩ trái [80] và truyền furosemide cải thiện trao đổi khí, cho phép giảm giá trị PEEP [81].
Người ta đã báo cáo rằng việc bổ sung albumin vào liệu pháp furosemide ở bệnh nhân giảm protein máu với ARDS cải thiện đáng kể quá trình oxy hóa, với cân bằng dịch ròng âm tính lớn hơn và duy trì ổn định huyết động tốt hơn [82]. Trong khi tổn thương phế nang lan tỏa (DAD), dấu hiệu mô bệnh học của ARDS, chỉ có ở 45% bệnh nhân được khám nghiệm tử thi đáp ứng các tiêu chuẩn định nghĩa của Berlin [83], giá trị EVLW trên 15 mL/ Kg xác định bệnh nhân có tổn thương phế nang lan tỏa với 99 % giá trị tiên đoán dương [77]. Do đó, đánh giá EVLW và PVPI có thể hữu ích ở bệnh nhân suy hô hấp giảm oxy máu cấp và thâm nhiễm phổi để phân biệt ARDS với phù tim [84, 85] và hướng dẫn xử trí dịch [78]. Tiêu chuẩn vàng để đo EVLW là pha loãng với chất chỉ nhiệt – màu qua phổi – yêu cầu tiêm đồng thời qua catheter tĩnh mạch trung tâm của một chất chỉ thị lạnh và một chất chỉ thị so màu. Tuy nhiên, kỹ thuật phức tạp và tốn kém này đã được thay thế bằng phương pháp pha loãng nhiệt qua phổi cho phép ước tính EVLW từ thể tích toàn phần cuối tâm trương (GEDV) (Hình 34.3). Phải thừa nhận một số hạn chế đáng kể của phép đo EVLW: EVLW có thể được đánh giá thấp khi có tắc mạch phổi và tràn dịch màng phổi, trong khi nó có thể được đánh giá quá cao ở bệnh nhân cắt phổi. Hơn nữa, PEEP có ảnh hưởng phức tạp đến phép đo EVLW: mức PEEP cao có thể chèn ép các mạch phổi có thành mỏng, dẫn đến lượng nước trong phổi bị đánh giá thấp [86], trong khi việc huy động các phế nang xẹp phổi với sự suy giảm co mạch do thiếu oxy có thể có tác dụng ngược lại. Tuy nhiên, một nghiên cứu trên bệnh nhân ARDS đã chứng minh mối tương quan tốt giữa EVLW và trọng lượng phổi được đo bằng chụp cắt lớp vi tính, độc lập với mức PEEP [87].
Một thông số quan trọng khác để đánh giá căn nguyên của phù phổi là Ppao, với giá trị > 18 mmHg cho thấy căn nguyên do tim. Theo dõi PAOP yêu cầu đặt PAC và có thể giúp duy trì áp lực mạch phổi ở mức thấp, giảm thiểu phù phổi và suy thành mao mạch do căng thẳng. Tuy nhiên, nhiều yếu tố (chủ yếu là rối loạn chức năng mạch máu, giảm sự tuân thủ của tâm thất và tăng ITP) có thể ảnh hưởng đến độ tin cậy của các giá trị đo được của CVP và PAOP như là các chỉ số về thể tích thất cuối tâm trương (tức là tiền tải thực tế của tim). Hơn nữa, ở bệnh nhân ARDS, hai hạn chế khác của phép đo Ppao phải được thừa nhận:
Tăng ITP do thông khí á lực dương, như đã báo cáo ở trên, gây ra sự gia tăng PAOP là do truyền Paw đến các mạch máu. Paw có thể được ước tính định lượng bằng cách sử dụng các phương pháp sau:
Ngắt máy thở tạm thời (nhưng mất PEEP đột ngột có thể gây xẹp phổi ở bệnh nhân ARDS nặng).
Chỉ số truyền: Teboul và các cộng sự đề xuất rằng tỷ lệ truyền áp lực phế nang đến mạch máu phổi có thể được tính bằng chỉ số truyền IT = (Ppao cuối kỳ thở vào – Ppao cuối kỳ thở ra)/ (Pplat – PEEP tổng). Do đó, Ppao xuyên thành có thể được tính là Ppao cuối kỳ thở ra – (IT × PEEP) [88].

Hình 34.3. Đo nước phổi ngoài mạch bằng cách pha loãng chỉ thị nhiệt đơn (thời gian vận chuyển trung bình MTt, thời gian giảm Dt).
Ppao có thể đánh giá thấp áp lực mao mạch phổi thực sự vì sức cản tĩnh mạch phổi tăng lên đáng kể và không thể bỏ qua [89]. Hai phương pháp đã được đề xuất để ước tính áp suất thủy tĩnh thực tế trong mao mạch phổi:
Phương trình Gaar: giả sử sức cản tĩnh mạch bằng khoảng 40% tổng sức cản mạch máu phổi, áp lực mao mạch hiệu dụng được tính là Ppc = Ppao + 0,4 × (mPAP – Ppao) [89, 90]. Tuy nhiên, phương pháp này không chính xác khi có những thay đổi đáng kể về sức cản mạch máu phổi.
Phân tích độ dốc của đường cong áp suất phân rã sau khi bơm bóng của PAC [91].
Cuối cùng, siêu âm phổi có thể được sử dụng để đánh giá mức độ phù phổi ở bệnh nhân ARDS: Lichtenstein và đồng nghiệp báo cáo rằng siêu âm phổi chính xác hơn X quang phổi khi so sánh với chụp cắt lớp vi tính. Hơn nữa, những phát hiện siêu âm phổi được đưa vào một trong những tiêu chuẩn chẩn đoán trong việc sửa đổi định nghĩa Berlin được đề xuất gần đây của Kigali [92].
Để đánh giá ảnh hưởng của việc truyền dịch lên chức năng phổi và đánh giá rủi ro và lợi ích của việc định vị PAC để hướng dẫn quản lý dịch truyền, Mạng lưới Thử nghiệm Lâm sàng ARDS đã thực hiện một nghiên cứu ngẫu nhiên lớn trên 1000 bệnh nhân ARDS được thở máy (Thử nghiệm điều trị bằng dịch truyền và catheter – FACTT) [93]. Dựa trên áp lực nội mạch đo được (CVP hoặc PAOP), bệnh nhân được chỉ định áp dụng phương pháp truyền dịch bảo tồn hoặc phương pháp truyền dịch tự do. Không quan sát thấy sự khác biệt về tỷ lệ tử vong trong 60 ngày, nhưng chiến lược bảo tồn đã rút ngắn thời gian thở máy và thời gian nằm ICU mà không làm tăng suy cơ quan ngoài phổi và cải thiện đáng kể chỉ số oxy và điểm tổn thương phổi. Một phân tích hậu kiểm của thử nghiệm này cho thấy rằng cả giảm oxy máu và chiến lược truyền dịch bảo tồn đều có liên quan độc lập với suy giảm chức năng tâm thần kinh lâu dài: vì không có bằng chứng về giảm tưới máu não (ví dụ, chỉ số tim, huyết áp tâm thu), nên vẫn chưa rõ mức độ bảo tồn trong quản lý dịch truyền có thể đã gây ra suy giảm nhận thức như thế [94]. Hơn nữa, không có sự khác biệt về tỷ lệ mắc hoặc thời gian của bất kỳ loại suy cơ quan nào cũng như nhu cầu hỗ trợ cơ quan (thuốc vận mạch, thông khí, thay thế thận) giữa liệu pháp truyền dịch có hướng dẫn bằng PAC và tĩnh mạch trung tâm; ngược lại, nhóm PAC có số biến chứng liên quan đến catheter cao gần gấp đôi. Tuy nhiên, catheter Swan-Ganz là một công cụ chẩn đoán, không phải là phương pháp điều trị [95], và kết quả FACTT chỉ ra rằng việc hồi sức tích cực bằng dịch truyền chứ không phải việc sử dụng PAC là nguyên nhân gây ra kết cục bất lợi. Cuối cùng, tăng cường thanh thải phù phổi bằng thuốc lợi tiểu, lọc máu hoặc bổ sung albumin giúp trao đổi khí tốt hơn và giảm thời gian thở máy ở bệnh nhân ARDS. Tuy nhiên, điều quan trọng ở bệnh nhân ARDS, mục tiêu tối ưu hóa DO2 có thể trái ngược với nhu cầu hạn chế dịch, đặc biệt khi ARDS xảy ra trong bối cảnh sốc và viêm toàn thân (ví dụ, nhiễm trùng huyết nặng, viêm tụy, bỏng). Trong những tình huống khó khăn này, việc tránh những tác động có hại của việc giảm DO2 và bảo tồn tưới máu các cơ quan chắc chắn cấp thiết hơn là “giữ cho phổi khô” [96, 97], và việc hồi sức tích cực bằng dịch truyền có thể cần thiết, chủ yếu được hướng dẫn bằng cách theo dõi CO và các chỉ số của tưới máu mô (như lactat và bão hòa tĩnh mạch trộn).
Chiến lược thông khí bảo vệ thất phải.
Trong thời đại thông khí bảo vệ, tần suất ACP đã giảm xuống, nhưng vẫn còn khoảng 20–25% [98]. Một số nghiên cứu chứng minh mối liên quan rõ ràng giữa cài đặt thở máy và tần suất ACP. Trong một nghiên cứu hồi cứu trên 352 bệnh nhân, Jardin đã tìm thấy mối quan hệ tuyến tính giữa tần suất ACP và giá trị Pplat: ACP được chẩn đoán ở 20% bệnh nhân có giá trị Pplat dưới 26 cmH2O, ở 39% những người có Pplat 27–35 cmH2O , và ở 42% bệnh nhân khi Pplat vượt quá 35 cmH2O [99]. Vì Pplat là một đại diện cho áp lực xuyên phổi, những phát hiện này xác nhận tác động có hại của việc tăng stress phổi đối với hậu tải RV. Các nghiên cứu gần đây cho thấy rằng áp lực đẩy (ΔPrs: chênh lệch thể tích thông khí giữa Pplat và PEEP, bằng tỷ lệ giữa thể tích thông khí trên tổng mức độ tuân thủ của hệ thống hô hấp) là một chỉ số tốt hơn về stress phổi [100, 101] và có liên quan độc lập với tỷ lệ tử vong và ACP [ 25]. Nó đã được chứng minh rằng nằm sấp có tác dụng có lợi trên huyết động bằng cách giảm tải của RV [102]; thực sự, bệnh nhân ARDS nặng nằm sấp trong thử nghiệm PROSEVA có suy tim mạch thấp hơn và ít ngừng tim hơn so với những bệnh nhân trong nhóm chứng [103]. Tư thế nằm sấp cho phép huy động phế nang đáng kể, cải thiện cả compliance của phổi (giảm áp lực xuyên phổi) và oxy hóa: những tác động này làm giảm đáng kể hậu tải RV và hậu quả là RV dãn rộng và rối loạn vận động vách liên thất cũng giảm đi [102]. Người ta đã đề xuất “điều gì tốt cho phổi thì tốt cho tâm thất phải và ngược lại” [5] nghĩa là cài đặt thở máy có thể được hướng dẫn bởi đánh giá chức năng RV. Đặc biệt, oxy vẫn là điểm quan trọng để bảo vệ RV, vì tỷ lệ PaO2/ FiO2 <100 mmHg là một yếu tố nguy cơ độc lập đối với ACP. Thể tích thông khí và PEEP nên được điều chỉnh để duy trì Pplat <27 cmH2O và áp lực đẩy <18 cmH2O, trong khi PaCO2 nên được duy trì
CÁC KẾT LUẬN.
Theo dõi chức năng hệ thống tim mạch có tầm quan trọng cơ bản ở bệnh nhân ARDS hoặc để xác định chẩn đoán hội chứng hoặc để phát hiện bất kỳ suy giảm huyết động nào có thể xảy ra. Việc tối ưu hóa cung cấp oxy, cung lượng tim và chức năng phổi đòi hỏi quản lý dịch truyền thận trọng và chiến lược thở máy cẩn thận.
Một nhóm chuyên gia gần đây đã xem xét các nguyên tắc theo dõi và quản lý huyết động ở bệnh nhân ARDS [105]: khuyến nghị đánh giá PPV ở bệnh nhân thụ động để đánh giá khả năng đáp ứng dịch và ảnh hưởng huyết động của thở máy. Ngoài ra, đề nghị đặt một catheter tĩnh mạch trung tâm để theo dõi Pra và ScVO2 và kiểm tra siêu âm tim thường xuyên để đánh giá chức năng RV và ước tính cung lượng tim. Trong những trường hợp phức tạp, khuyến nghị đặt PAC hoặc sử dụng hệ thống pha loãng nhiệt xuyên phổi. Thuật toán này chắc chắn có một cơ sở lý luận mạnh mẽ. Tuy nhiên, chúng tôi muốn nhấn mạnh rằng, đặc biệt trong những trường hợp phức tạp, PAC vẫn là một công cụ chẩn đoán cần thiết ở những bệnh nhân ARDS nặng, vì nó là thiết bị duy nhất cho phép theo dõi liên tục áp lực động mạch phổi, áp lực đổ đầy tim và SVO2 “thực tế”; ngoài ra, nó còn cho phép đo cung lượng tim [106]. Những thông tin này rất quan trọng đối với cả việc tối ưu hóa huyết động và thiết lập thông khí thông qua thở máy, để cải thiện việc cung cấp oxy đồng thời tránh rối loạn chức năng tim phải [104].
Thông tin cốt lõi.
Tăng áp động mạch phổi và rối loạn chức năng thất phải ảnh hưởng tiêu cực đến tiên lượng của bệnh nhân ARDS. Tránh quá tải dịch và giảm áp lực căng phổi trong quá trình thở máy là những chiến lược hợp lý để bảo toàn cung lượng tim.
Tối ưu hóa cung cấp oxy có vai trò quan trọng để ngăn ngừa sự suy giảm năng lượng sinh học của các mô cơ thể: về mặt này, đánh giá tình trạng thể tích nội mạch và khả năng đáp ứng dịch truyền là rất quan trọng để hướng dẫn hỗ trợ huyết động đồng thời tránh quá tải dịch.
Do sự cân bằng mong manh giữa nhu cầu điều trị tim và phổi, nên theo dõi huyết động cẩn thận là bắt buộc: PAC, pha loãng nhiệt qua phổi (ví dụ: công nghệ PiCCO hoặc Vigileo) và siêu âm tim cung cấp thông tin hữu ích cho sự thay đổi nhanh chóng và linh hoạt trong quá trình ra quyết định y tế.
TÀI LIỆU THAM KHẢO.
Lorente JA, Ballén-Barragán A, Herrero R, Esteban A. Acute respiratory distress syndrome: does histology matter? Crit Care. 2015;19: 337.
Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt 1): 818–24.
Definition Task Force ARDS, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23): 2526–33.
Vieillard-Baron A, Aneman A. Cardiovascular issues in the ICU: a call for papers. Intensive Care Med. 2017;43(12): 1892–3.
Repessé X, Charron C, Vieillard-Baron A. Acute respiratory distress syndrome: the heart side of the moon. Curr Opin Crit Care. 2016;22(1): 38–44.
Vieillard-Baron A, Girou E, Valente E, Brun-Buisson C, Jardin F, Lemaire F, Brochard L. Predictors of mortality in acute respiratory distress syndrome. Focus On the role of right heart catheterization. Am J Respir Crit Care Med. 2000;161(5): 1597–601.
Davies SW, Wedzicha JA. Hypoxia and the heart. Br Heart J. 1993;69(1): 3–5.
Poole-Wilson PA. Acidosis and contractility of heart muscle. Ciba Found Symp. 1982;87: 58– 76.
Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, Ranieri M, Rubenfeld G, Thompson BT, Wrigge H, Slutsky AS, Pesenti A, LUNG SAFE Investigators; ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8): 788–800.
Zapol WM, Snider MT. Pulmonary hypertension in severe acute respiratory failure. N Engl J Med. 1977;296(9): 476–80.
Zapol WM, Kobayashi K, Snider MT, Greene R, Laver MB. Vascular obstruction causes pulmonary hypertension in severe acute respiratory failure. Chest. 1977;71(2 suppl): 306–7.
Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC); European Respiratory Society (ERS); International Society of Heart and Lung Transplantation (ISHLT), Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6): 1219–63.
Osman D, Monnet X, Castelain V, Anguel N, Warszawski J, Teboul JL, Richard C. French Pulmonary Artery Catheter Study Group. Incidence and prognostic value of right ventricular failure in acute respiratory distress syndrome. Intensive Care Med. 2009;35(1): 69–76.
Villar J, Blazquez MA, Lubillo S, Quintana J, Manzano JL. Pulmonary hypertension in acute respiratory failure. Crit Care Med. 1989;17(6): 523–6.
Cepkova M, Kapur V, Ren X, Quinn T, Zhuo H, Foster E, Liu KD, Matthay MA. Pulmonary dead space fraction and pulmonary artery systolic pressure as early predictors of clinical outcome in acute lung injury. Chest. 2007;132(3): 836–42.
Bull TM, Clark B, McFann K, Moss M, National Institutes of Health/National Heart, Lung, and Blood Institute ARDS Network. Pulmonary vascular dysfunction is associated with poor outcomes in patients with acute lung injury. Am J Respir Crit Care Med. 2010;182(9): 1123–8.
Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30(4): 394– 403.
Ryan D, Frohlich S, McLoughlin P. Pulmonary vascular dysfunction in ARDS. Ann Intensive Care. 2014;4: 28.
Calcaianu G, Calcaianu M, Gschwend A, Canuet M, Meziani F, Kessler R. Hemodynamic profile of pulmonary hypertension (PH) in ARDS. Pulm Circ. 2018;8(1):204589321775341.
Versprille A. Pulmonary vascular resistance. A meaningless variable. Intensive Care Med. 1984;10(2): 51–3.
Naeije R. Pulmonary vascular resistance. A meaningless variable? Intensive Care Med. 2003;29(4): 526–9.
Petersson J, Ax M, Frey J, Sánchez-Crespo A, Lindahl SG, Mure M. Positive end-expiratory pressure redistributes regional blood flow and ventilation differently in supine and prone humans. Anesthesiology. 2010;113(6): 1361–9.
Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol (1985). 2005;98(1): 390–403.
Repessé X, Charron C, Vieillard-Baron A. Assessment of the effects of inspiratory load on right ventricular function. Curr Opin Crit Care. 2016;22(3): 254–9.
Boissier F, Katsahian S, Razazi K, Thille AW, Roche-Campo F, Leon R, Vivier E, Brochard L, VieillardBaron A, Brun-Buisson C, Mekontso Dessap A. Prevalence and prognosis of cor pulmonale during protective ventilation for acute respiratory distress syndrome. Intensive Care Med. 2013;39(10): 1725–33.
Lhéritier G, Legras A, Caille A, Lherm T, Mathonnet A, Frat JP, Courte A, Martin-Lefèvre L, Gouëllo JP, Amiel JB, Garot D, Vignon P. Prevalence and prognostic value of acute cor pulmonale and patent foramen ovale in ventilated patients with early acute respiratory distress syndrome: a multicenter study. Intensive Care Med. 2013;39(10): 1734–42.
Price LC, McAuley DF, Marino PS, Finney SJ, Griffiths MJ, Wort SJ. Pathophysiology of pulmonary hypertension in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302(9): L803–15.
Tomashefski JF Jr, Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol. 1983;112(1):112–26.
Ware LB, Koyama T, Zhao Z, Janz DR, Wickersham N, Bernard GR, May AK, Calfee CS, Matthay MA. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care. 2013;17(5): R253.
West JB. Regional differences in the lung. Chest. 1978;74(4): 426–37.
Whittenberger JL, McGregor M, Berglund E, Borst HG. Influence of state of inflation of the lung on pulmonary vascular resistance. J Appl Physiol. 1960;15: 878–82.
Permutt S, Bromberger-Barnea B, Bane HN. Alveolar pressure, pulmonary venous pressure, and the vascular waterfall. Med Thorac. 1962;19: 239–60.
Jardin F. Ventricular interdependence: how does it impact on hemodynamic evaluation in clinical practice? Intensive Care Med. 2003;29(3):361–3.
Wauthy P, Pagnamenta A, Vassalli F, Naeije R, Brimioulle S. Right ventricular adaptation to pulmonary hypertension: an interspecies comparison. Am J Physiol Heart Circ Physiol. 2004;286(4):H1441.
Mitchell SC, Lelieveldt BP, van der Geest RJ, Bosch HG, Reiber JH, Sonka M. Multistage hybrid active appearance model matching: segmentation of left and right ventricles in cardiac MR images. IEEE Trans Med Imaging. 2001;20(5):415–23.
Zochios V, Parhar K, Tunnicliffe W, Roscoe A, Gao F. The right ventricle in ARDS. Chest. 2017;152(1): 181–93.
Repessé X, Charron C, Vieillard-Baron A. Acute cor pulmonale in ARDS: rationale for protecting the right ventricle. Chest. 2015;147(1): 259–65.
Feihl F, Broccard AF. Interactions between respiration and systemic hemodynamics. Part I: basic concepts. Intensive Care Med. 2009;35(1): 45–54.
Marini JJ, Ravenscraft SA. Mean airway pressure: physiologic determinants and clinical importance-Part 1: physiologic determinants and measurements. Crit Care Med. 1992;20(10): 1461–72.
Qvist J, Pontoppidan H, Wilson RS, Lowenstein E, Laver MB. Hemodynamic responses to mechanical ventilation with PEEP: the effect of hypervolemia. Anesthesiology. 1975;42(1): 45–55.
Nanas S, Magder S. Adaptations of the peripheral circulation to PEEP. Am Rev Respir Dis. 1992;146(3): 688–93.
Maas J, Lagrand WK, Van den Berg PC, Pinsky MR, Jansen JR. Venous return in ICU patients. Crit Care. 2008;12(Suppl 2): P93.
Jellinek H, Krenn H, Oczenski W, Veit F, Schwarz S, Fitzgerald RD. Influence of positive airway pressure on the pressure gradient for venous return in humans. J Appl Physiol (1985). 2000;88(3): 926–32.
Marini JJ, Culver BH, Butler J. Mechanical effect of lung distention with positive pressure on cardiac function. Am Rev Respir Dis. 1981;124(4): 382–6.
Cherpanath TG, Lagrand WK, Schultz MJ, Groeneveld AB. Cardiopulmonary interactions during mechanical ventilation in critically ill patients. Neth Heart J. 2013;21(4): 166–72.
Bendjelid K, Schütz N, Suter PM, Romand JA. Continuous cardiac output monitoring after cardiopulmonary bypass: a comparison with bolus thermodilution measurement. Intensive Care Med. 2006;32(6): 919–22.
Chapin JC, Downs JB, Douglas ME, Murphy EJ, Ruiz BC. Lung expansion, airway pressure transmission, and positive end-expiratory pressure. Arch Surg. 1979;114(10): 1193–7.
Deal C, Osborn JJ, Ellis E, Gerbode F. Chest wall compliance. Ann Surg. 1968;167(1): 73–7.
Brochard L, Slutsky A, Pesenti A. Mechanical ventilation to minimize progression of lung injury in acute respiratory failure. Am J Respir Crit Care Med. 2017;195(4): 438–42.
Bellani G, Grasselli G, Teggia-Droghi M, Mauri T, Coppadoro A, Brochard L, Pesenti A. Do spontaneous and mechanical breathing have similar effects on average transpulmonary and alveolar pressure? A clinical crossover study. Crit Care. 2016;20(1): 142.
Mauri T, Cambiaghi B, Spinelli E, Langer T, Grasselli G. Spontaneous breathing: a double-edged sword to handle with care. Ann Transl Med. 2017;5(14): 292. https://doi.org/10.21037/atm.2017.06.55.
ELSO Guidelines 2017.
Netzer G, Shah CV, Iwashyna TJ, Lanken PN, Finkel B, Fuchs B, Guo W, Christie JD. Association of RBC transfusion with mortality in patients with acute lung injury. Chest. 2007;132(4): 1116–23.
Gong MN, Thompson BT, Williams P, Pothier L, Boyce PD, Christiani DC. Clinical predictors of and mortality in acute respiratory distress syndrome: potential role of red cell transfusion. Crit Care Med. 2005;33(6): 1191–8.
Gaggar A, Patel RP. There is blood in the water: hemolysis, hemoglobin, and heme in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2016;311(4): L714.
Alhashemi JA, Cecconi M, Hofer CK. Cardiac output monitoring: an integrative perspective. Crit Care. 2011;15(2): 214.
Mercado P, Maizel J, Beyls C, Titeca-Beauport D, Joris M, Kontar L, Riviere A, Bonef O, Soupison T, Tribouilloy C, de Cagny B, Slama M. Transthoracic echocardiography: an accurate and precise method for estimating cardiac output in the critically ill patient. Crit Care. 2017;21(1): 136.
Vincent JL, Weil MH. Fluid challenge revisited. Crit Care Med. 2006;34(5): 1333–7.
Carsetti A, Cecconi M, Rhodes A. Fluid bolus therapy: monitoring and predicting fluid responsiveness. Curr Opin Crit Care. 2015;21(5): 388–94.
Coudray A, Romand JA, Treggiari M, Bendjelid K. Fluid responsiveness in spontaneously breathing patients: a review of indexes used in intensive care. Crit Care Med. 2005;33(12): 2757–62.
Osman D, Ridel C, Ray P, Monnet X, Anguel N, Richard C, Teboul JL. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med. 2007;35(1): 64–8.
Marik PE. Noninvasive cardiac output monitors: a state-of the-art review. J Cardiothorac Vasc Anesth. 2013;27(1): 121–34.
Magder S. Central venous pressure monitoring. Curr Opin Crit Care. 2006;12(3): 219–27.
Hasanin A. Fluid responsiveness in acute circulatory failure. J Intensive Care. 2015;3: 50.
Monnet X, Rienzo M, Osman D, Anguel N, Richard C, Pinsky MR, Teboul JL. Passive leg raising predicts fluid responsiveness in the critically ill. Crit Care Med. 2006;34(5): 1402–7.
Saugel B, Bendjelid K, Critchley LAH, Scheeren TWL. Journal of Clinical Monitoring and Computing 2017 end of year summary: cardiovascular and hemodynamic monitoring. J Clin Monit Comput. 2018;32(2): 189–96.
Zhang D, Song Y, Yang Y, Duan A, Zhang Z, Wang Y. An application of arterial pressurebased cardiac output measurements in fluid management strategies of critically ill patients. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2014;26(9): 620–3.
Teboul JL. Mean systemic pressure: we can now estimate it, but for what? Intensive Care Med. 2013;39(8): 1487–8.
Lefrant JY, De Backer D. Can we use pulse pressure variations to predict fluid responsiveness in patients with ARDS? Intensive Care Med. 2009;35(6): 966–8.
Silva S, Jozwiak M, Teboul JL, Persichini R, Richard C, Monnet X. End-expiratory occlusion test predicts preload responsiveness independently of positive end-expiratory pressure during acute respiratory distress syndrome. Crit Care Med. 2013;41(7): 1692–701.
Mezger V, Balzer F, Habicher M, Sander M. Venous saturation: between oxygen delivery and consumption. Med Klin Intensivmed Notfmed. 2017;112(6): 492–8.
López A, Grignola JC, Angulo M, Alvez I, Nin N, Lacuesta G, Baz M, Cardinal P, Prestes I, Bouchacourt JP, Riva J, Ince C, Hurtado FJ. Effects of early hemodynamic resuscitation on left ventricular performance and microcirculatory function during endotoxic shock. Intensive Care Med Exp. 2015;3(1): 49.
Sakr Y, Rubatto Birri PN, Kotfis K, Nanchal R, Shah B, Kluge S, Schroeder ME, Marshall JC, Vincent JL, Intensive Care Over Nations Investigators. Higher fluid balance increases the risk of death from sepsis: results from a large international audit. Crit Care Med. 2017;45(3): 386– 94.
Bagshaw SM, Brophy PD, Cruz D, Ronco C. Fluid balance as a biomarker: impact of fluid overload on outcome in critically ill patients with acute kidney injury. Crit Care. 2008;12(4): 169.
Alobaidi R, Morgan C, Basu RK, Stenson E, Featherstone R, Majumdar SR, Bagshaw SM. Association between fluid balance and outcomes in critically ill children: a systematic review and meta-analysis. JAMA Pediatr. 2018;172(3): 257–68.
Tagami T, Ong MEH. Extravascular lung water measurements in acute respiratory distress syndrome: why, how, and when? Curr Opin Crit Care. 2018;24(3): 209–15.
Tagami T, Sawabe M, Kushimoto S, Marik PE, Mieno MN, Kawaguchi T, Kusakabe T, Tosa R, Yokota H, Fukuda Y. Quantitative diagnosis of diffuse alveolar damage using extravascular lung water. Crit Care Med. 2013;41(9): 2144–50.
Jozwiak M, Teboul JL, Monnet X. Extravascular lung water in critical care: recent advances and clinical applications. Ann Intensive Care. 2015;5(1): 38.
Gattinoni L, Cressoni M, Brazzi L. Fluids in ARDS: from onset through recovery. Curr Opin Crit Care. 2014;20(4): 373–7.
Prewitt RM, McCarthy J, Wood LD. Treatment of acute low pressure pulmonary edema in dogs: relative effects of hydrostatic and oncotic pressure, nitroprusside, and positive endexpiratory pressure. J Clin Invest. 1981;67(2): 409–18.
Reising CA, Chendrasekhar A, Wall PL, Paradise NF, Timberlake GA, Moorman DW. Continuous dose furosemide as a therapeutic approach to acute respiratory distress syndrome (ARDS). J Surg Res. 1999;82(1): 56–60.
Martin GS, Moss M, Wheeler AP, Mealer M, Morris JA, Bernard GR. A randomized, controlled trial of furosemide with or without albumin in hypoproteinemic patients with acute lung injury. Crit Care Med. 2005;33(8): 1681–7.
Thille AW, Esteban A, Fernández-Segoviano P, Rodriguez JM, Aramburu JA, Peñuelas O, Cortés-Puch I, Cardinal-Fernández P, Lorente JA, Frutos-Vivar F. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am J Respir Crit Care Med. 2013;187(7): 761–7.
Kushimoto S, Taira Y, Kitazawa Y, Okuchi K, Sakamoto T, Ishikura H, Endo T, Yamanouchi S, Tagami T, Yamaguchi J, Yoshikawa K, Sugita M, Kase Y, Kanemura T, Takahashi H, Kuroki Y, Izumino H, Rinka H, Seo R, Takatori M, Kaneko T, Nakamura T, Irahara T, Saito N, Watanabe A, PiCCO Pulmonary Edema Study Group. The clinical usefulness of extravascular lung water and pulmonary vascular permeability index to diagnose and characterize pulmonary edema: a prospective multicenter study on the quantitative differential diagnostic definition for acute lung injury/acute respiratory distress syndrome. Crit Care. 2012;16(6): R232.
Perel A. Extravascular lung water and the pulmonary vascular permeability index may improve the definition of ARDS. Crit Care. 2013;17(1):108.
Carlile PV, Beckett RC, Gray BA. Relationship between CO and transit times for dye and thermal indicators in central circulation. J Appl Physiol (1985). 1986;60(4): 1363–72.
Patroniti N, Bellani G, Maggioni E, Manfio A, Marcora B, Pesenti A. Measurement of pulmonary edema in patients with acute respiratory distress syndrome. Crit Care Med. 2005;33(11): 2547–54.
Teboul JL, Pinsky MR, Mercat A, Anguel N, Bernardin G, Achard JM, Boulain T, Richard C. Estimating cardiac filling pressure in mechanically ventilated patients with hyperinflation. Crit Care Med. 2000;28(11): 3631–6.
Ganter CC, Jakob SM, Takala J. Pulmonary capillary pressure. A review. Minerva Anestesiol. 2006;72(1– 2):21–36. Review Erratum in: Minerva Anestesiol 2007 Mar;73(3): XVII.
Gaar KA J, Taylor AE, Owens LJ, Guyton AC. Pulmonary capillary pressure and filtration coefficient in the isolated perfused lung. Am J Phys. 1967;213(4): 910–4.
Takala J. Pulmonary capillary pressure. Intensive Care Med. 2003;29(6): 890–3.
Riviello ED, Buregeya E, Twagirumugabe T. Diagnosing acute respiratory distress syndrome in resource limited settings: the Kigali modification of the Berlin definition. Curr Opin Crit Care. 2017;23(1): 18–23.
National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, de Boisblanc B, Connors AF Jr, Hite RD, Harabin AL. Comparison of two fluidmanagement strategies in acute lung injury. N Engl J Med. 2006;354(24): 2564–75.
Mikkelsen ME, Christie JD, Lanken PN, Biester RC, Thompson BT, Bellamy SL, Localio AR, Demissie E, Hopkins RO, Angus DC. The adult respiratory distress syndrome cognitive outcomes study: longterm neuropsychological function in survivors of acute lung injury. Am J Respir Crit Care Med. 2012;185(12): 1307–15.
Chatterjee K. The Swan-Ganz catheters: past, present, and future. A viewpoint. Circulation. 2009;119(1): 147–52.
Levy MM, Artigas A, Phillips GS, Rhodes A, Beale R, Osborn T, Vincent JL, Townsend S, Lemeshow S, Dellinger RP. Outcomes of the surviving sepsis campaign in intensive care units in the USA and Europe: a prospective cohort study. Lancet Infect Dis. 2012;12(12): 919–24.
Tenner S, Baillie J, DeWitt J, Vege SS, American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108(9): 1400–15.
Vieillard-Baron A, Schmitt JM, Augarde R, Fellahi JL, Prin S, Page B, Beauchet A, Jardin F. Acute cor pulmonale in acute respiratory distress syndrome submitted to protective ventilation: incidence, clinical implications, and prognosis. Crit Care Med. 2001;29(8): 1551–5.
Jardin F, Vieillard-Baron A. Right ventricular function and positive pressure ventilation in clinical practice: from hemodynamic subsets to respirator settings. Intensive Care Med. 2003;29(9): 1426–34.
Mauri T, Lazzeri M, Bellani G, Zanella A, Grasselli G. Respiratory mechanics to understand ARDS and guide mechanical ventilation. Physiol Meas. 2017;38(12): R280–H303.
Amato MB, Meade MO, Slutsky AS, Brochard L, Costa EL, Schoenfeld DA, Stewart TE, Briel M, Talmor D, Mercat A, Richard JC, Carvalho CR, Brower RG. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med. 2015;372(8): 747–55.
Vieillard-Baron A, Charron C, Caille V, Belliard G, Page B, Jardin F. Prone positioning unloads the right ventricle in severe ARDS. Chest. 2007;132(5): 1440–6.
Guérin C, Reignier J, Richard JC, Beuret P, Gacouin A, Boulain T, Mercier E, Badet M, Mercat A, Baudin O, Clavel M, Chatellier D, Jaber S, Rosselli S, Mancebo J, Sirodot M, Hilbert G, Bengler C, Richecoeur J, Gainnier M, Bayle F, Bourdin G, Leray V, Girard R, Baboi L, Ayzac L, PROSEVA Study Group. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368(23): 2159–68.
Paternot A, Repessé X, Vieillard-Baron A. Rationale and description of right ventricleprotective ventilation in ARDS. Respir Care. 2016;61(10): 1391–6.
Vieillard-Baron A, Matthay M, Teboul JL, Bein T, Schultz M, Magder S, Marini JJ. Experts’ opinion on management of hemodynamics in ARDS patients: focus on the effects of mechanical ventilation. Intensive Care Med. 2016;42(5): 739–49.
Ventetuolo CE, Klinger JR. Management of acute right ventricular failure in the intensive care unit. Ann Am Thorac Soc. 2014;11: 811–22.
BÌNH LUẬN