Sự hiểu biết của chúng ta về lịch sử tự nhiên của bệnh thận tiểu đường đã xuất hiện phần lớn từ những bệnh nhân mắc bệnh tiểu đường loại 1. Tuy nhiên, các biểu hiện mô học ở những người mắc bệnh tiểu đường loại 2 là tương tự nhau. Cả biểu hiện lâm sàng và sự xuất hiện mô học của bệnh thận liên quan đến bệnh tiểu đường đã được hiểu tương đối rõ ràng. Tuy nhiên, sinh bệnh học ít được hiểu rõ hơn và có những khoảng trống trong sự hiểu biết của chúng ta về cách các yếu tố nguyên nhân khác nhau liên quan đến các biểu hiện mô học của bệnh tiểu đường; Một phần của điều này là do sự ít ỏi của sinh thiết thận và dữ liệu dọc. Ở đây, chúng tôi sẽ tập trung vào sinh bệnh học, tóm tắt sự hiểu biết hiện tại của chúng ta về mối tương quan mô học và lâm sàng và chỉ ra những tranh cãi còn lại trong bối cảnh sinh bệnh học.
Cơ chế bệnh sinh của bệnh thận do tiểu đường được bắt đầu và duy trì bởi bốn yếu tố nguyên nhân, có thể được phân loại thành các yếu tố chuyển hóa, huyết động, tăng trưởng và các yếu tố tiền viêm hoặc xơ hóa (Hình 1). Mặc dù có sự chồng chéo đáng kể giữa các yếu tố này và sự thay đổi trong đóng góp tương đối của chúng giữa các cá nhân và theo thời gian, để dễ thảo luận, chúng tôi sẽ mô tả sinh bệnh học như thể mỗi yếu tố đóng một vai trò riêng biệt. Những yếu tố gây bệnh này tạo ra các tổn thương ở các khoang thận khác nhau: cầu thận, ống thận, mô kẽ và mạch máu. Một loạt các phân tử, thụ thể, enzyme và các yếu tố phiên mã phức tạp tham gia vào quá trình thúc đẩy giai đoạn sớm nhất của bệnh thận đến thận to với phì đại, ma trận ngoại bào mở rộng (ECM), xơ cứng cầu thận, thoái hoá hialine mạch máu, xơ hóa mô kẽ và teo ống thận, và mất chức năng dẫn đến đỉnh điểm là bệnh thận giai đoạn cuối (ESRD).
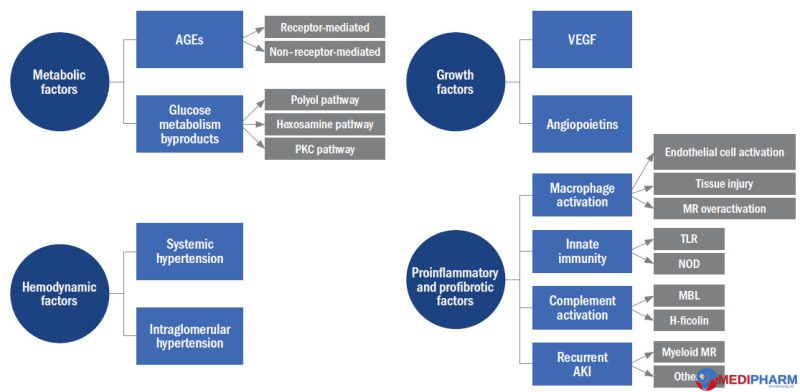
Hình 1. Tổng quan về các yếu tố gây bệnh trong bệnh thận đái tháo đường. Các thành tố điều khiển chính của bệnh thận tiểu đường có thể được phân loại là các yếu tố chuyển hóa, huyết động, tăng trưởng và các yếu tố tiền viêm hoặc xơ hóa.
Yếu tố chuyển hoá
Những thay đổi sớm nhất được kích hoạt bởi các yếu tố chuyển hoá, cụ thể là tăng đường huyết. Tốn hại do tăng đường huyết có thể xảy ra do thay đổi các mô hoặc có thể được gây ra bởi các sản phẩm chuyển hóa glucose (11). Tổng quan về các rối loạn con đường chuyển hoá làm trung gian sinh bệnh học của bệnh thận ở những người mắc bệnh tiểu đường được thể hiện trong Hình 2.

Hình 2. Con đường chuyển hoá của bệnh thận tiểu đường. Tăng đường huyết kích thích sự tích tụ của AGEs và các sản phẩm khác của chuyển hóa glucose. Kích hoạt từng con đường này có thể làm tổn thương thận. AGEs có thể tạo ra tổn thương tế bào bởi các con đường thụ thể và không thụ thể. Bên ngoài các tế bào, chúng có thể gây tổn thương mô bằng cách glycosyl hoá không enzym các phân tử như collagen có thể làm giảm sự thống nhất mô thông qua liên kết chéo. Tăng thông lượng glucose có thể dẫn đến kích hoạt các con đường như polyol, hexosamine và PKC có thể dẫn đến tổn thương tế bào và rối loạn chức năng cơ quan.
Sự Glycosyl hoá của các mô
Tăng đường huyết thông qua cơ chế không enzyme có thể dẫn đến sản xuất các sản phẩm đầu cuối bền vững của quá trình glycosyl hoá (AGEs), bằng cách Glycosyl hoá không enzym các thành phần mô khác nhau như protein, collagen, lipid và ECM có thể gây rối loạn chức năng cơ quan. Quá trình này được ví như quá trình lão hóa tăng tốc thông qua chuyển sang màu nâu của các mô hoặc phản ứng Maillard (11).
Glycosyl hoá không enzym của các phân tử gây ra tổn thương hạ lưu bởi một số cơ chế có thể được phân loại thành các loại qua trung gian thụ thể và không qua trung gian thụ thể (12).
Glycosyl hoá không enzym dẫn đến kích hoạt các thụ thể trên tế bào – đặc trưng tốt nhất trong số đó là thụ thể của các sản phẩm cuối bền vững của quá trình glycosyl hoá (RAGE) – kích hoạt sự tổng hợp và giải phóng yếu tố hạt nhân κB (NFκB) và tạo ra các loại oxy phản ứng (ROS). Các phân tử này, mặc dù các yếu tố phiên mã, bắt đầu và duy trì tổn thương thận bằng một số quá trình (12), bao gồm tăng trưởng tế bào và phì đại, viêm, tạo mạch, rối loạn chức năng nội mô và sản xuất ECM.
Trong các tế bào, AGEs có thể tạo ra rối loạn chức năng tế bào mà không liên kết với một thụ thể. Ví dụ, glycosyl hoá protein cytosolic có thể làm giảm khả dụng sinh học oxit nitric (NO) và gây ra stress oxy hóa (12). Tương tự, bên ngoài các tế bào, AGEs có thể gây ra rối loạn chức năng mô mà không liên kết với thụ thể. Ví dụ, sự Glycosyl hoá không enzym của các thành phần mô liên kết như collagen có thể liên kết chéo các phân tử trong ECM và gây rối loạn chức năng (12).
Các biểu hiện mô học của tích lũy AGE bao gồm dày màng đáy, giảm thoái hóa protein dẫn đến tăng ma trận trung mô và tăng thể tích ngoại bào kẽ.
Tổn hại gây ra bởi các sản phẩm chuyển hóa glucose
Glucose có thể gây ra tổn hại trong các tế bào độc lập với sự glycosyl hoá không enzym như bằng cách kích hoạt con đường polyol, con đường hexosamine hoặc con đường protein kinase C (PKC) hoặc thông qua việc tạo ra ROS.
Con đường Polyol
Con đường polyol liên quan đến việc kích hoạt enzyme aldose reductase trong tế bào khi nồng độ glucose nội bào tăng đến mức tăng đường huyết (11). Điều này làm cạn kiệt nồng độ nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) của tế bào và làm thay đổi tỷ lệ oxy hóa khử, có thể làm giảm NO sinh khả dụng và thay đổi chức năng enzyme. Mặc dù các chất ức chế aldose reductase đã được tìm thấy có hiệu quả trong các mô hình gặm nhấm của bệnh tiểu đường, các thử nghiệm trên người đã thất bại trong việc tiết lộ sự bảo vệ khỏi một biến chứng vi mạch quan trọng của bệnh tiểu đường – bệnh về mắt – trong một thử nghiệm ngẫu nhiên (13).
Con đường Hexosamine
Con đường hexosamine rất quan trọng cho sự tổng hợp proteoglycans, glycolipid và glycoprotein (14). Sự tổng hợp của các phân tử này đòi hỏi một chất nền đường amin gọi là UDP-N-acetylglucosamine, là sản phẩm cuối cùng của con đường hexosamine. Enzym giới hạn tốc độ của con đường hexosamine là glutamine: fructose-6-phosphate-amidotransferase (GFAT), xúc tác phản ứng giữa fructose-6-phosphate và glutamine cho amine để tạo ra glucosamine-6-phosphate (14). Trong các tế bào màng mạch cầu thận nuôi cấy, nồng độ glucose cao kích thích sản xuất yếu tố tăng trưởng biến đổi β1 (TGF-β1); tác dụng này được loại bỏ bằng cách ức chế GFAT. Ngược lại, sự biểu hiện quá mức ổn định của GFAT làm tăng sản xuất TGF-β1. Hơn nữa, các hiệu ứng dường như được chuyển đổi bởi PKC. Ở người, GFAT không có trong các tế bào cầu thận. Tuy nhiên, ở những bệnh nhân mắc bệnh thận tiểu đường, GFAT được biểu hiện trong cầu thận, cho thấy nó có thể đóng vai trò sinh lý bệnh (14).
Con đường PKC
PKC là một họ enzyme là các phân tử tín hiệu nội bào then chốt và rất quan trọng đối với chức năng mạch máu. Ở trạng thái sinh lý, kích hoạt PKC qua trung gian thụ thể giải phóng các ion canxi nội bào và diacylglycerol (DAG) và kích hoạt các enzyme này. Trong các trạng thái bệnh lý như trong bệnh tiểu đường, sản xuất DAG có thể tăng bất thường và có thể dẫn đến kích hoạt PKC. Trong bệnh tiểu đường, sản xuất DAG được tăng lên bằng cách tăng ly giải glycogen và mức độ glyceraldehyd-3-phosphate nội bào và glycerol-3-phosphate tăng cao. PKC cũng có thể được kích hoạt bởi ROS và AGEs. Một chất ức chế PKC-β ruboxistaurin đã được thử nghiệm trong một thử nghiệm lâm sàng ngẫu nhiên giai đoạn 2 ở bệnh nhân tiểu đường loại 2 và albumin niệu dai dẳng (tỷ lệ albumin trên creatinine [ACR] 200-2.000 mg / g creatinine) mặc dù điều trị bằng thuốc ức chế hệ thống renin-angiotensin (15). So với giả dược, việc giảm ACR sau 1 năm – điểm cuối chính của nghiên cứu – không đáng kể.
Các yếu tố huyết động
Sự gia tăng áp lực mao mạch cầu thận làm tăng tốc độ lọc cầu thận nephron đơn – siêu lọc – và điều này xảy ra sớm trong quá trình bệnh tiểu đường. Sự gia tăng áp lực trong cầu thận là kết quả của sự gia tăng trương lực tiểu động mạch đi và giảm trương lực tiểu động mạch đến (Hình 3) (16). Làm thế nào quá trình này xảy ra không được hiểu đầy đủ, nhưng hai lý thuyết đã xuất hiện.

Hình 3. Cơ chế tăng áp lực trong cầu thận. Áp lực trong cầu thận có thể tăng do tăng trương lực tiểu động mạch đi hoặc giảm trương lực tiểu động mạch đến. Các yếu tố trung gian của những thay đổi này được hiển thị.
Một nhóm tin rằng siêu lọc được điều chỉnh bởi các phân tử tuần hoàn chủ yếu hoạt động trong cầu thận (17). Một số chất trung gian đã được đề xuất cho tăng áp lực trong cầu thận thông qua việc tăng trương lực tiểu động mạch đi và giảm trương lực tiểu động mạch đến. Tăng sức cản tiểu động mạch đi có thể là kết quả của sự gia tăng nồng độ angiotensin II, thromboxane A2 (TxA2), endothelin 1 (ET-1) và ROS (16). Giảm sức cản tiểu động mạch đến có thể được kích thích bằng cách giảm sinh khả dụng NO oxide; tăng prostanoids cyclooxygenase-2 (COX-2); kích hoạt hệ thống kallikrein-kinin, peptide lợi niệu nhĩ và angiotensin 1-7; và tăng insulin (16).
Tuy nhiên, một nhóm khác đề xuất rằng các cơ chế ống thận vẫn là động lực chính của tăng áp lực trong cầu thận (12). Việc kích hoạt các con đường vận chuyển glucose trong ống lượn gần trong quá trình bệnh tiểu đường kích thích sự tái hấp thu cả glucose và natri trong nephron gần (12). Cung cấp natri cho nephron xa bị giảm. Điều này kích hoạt phản hồi ống-cầu thận; Tiểu động mạch đến giãn ra, và tiểu động mạch đi co lại (12). Sự gia tăng insulin tự nó có thể làm tăng vận chuyển natri và glucose trong ống thận gần và kích thích phản hồi ống cầu thận. Insulin, như đã lưu ý ở trên, cũng có thể làm giảm trương lực tiểu động mạch đến trực tiếp. Do đó, insulin có thể trực tiếp và gián tiếp gây ra siêu lọc.
Các yếu tố tăng trưởng
Từ lâu, người ta đã nhận ra rằng bệnh lý vi mạch như xảy ra trong mắt cũng liên quan đến bệnh thận. Do đó, các nhà điều tra đã khám phá mối quan hệ giữa tăng sinh mạch máu và tính thấm nội mô – các yếu tố được biết là quan trọng trong sinh bệnh học của bệnh mắt tiểu đường – với sự xuất hiện của bệnh thận tiểu đường. Yếu tố tăng trưởng nội mô mạch máu (VEGF) được kích hoạt sớm và dẫn đến giãn nở mạch máu, có thể gây ra xơ cứng hyaline động mạch và thay đổi tăng huyết áp ở thận (18). Tương tự, angiopoietin có thể gây tăng sinh mạch máu và có liên quan đến sinh bệnh học của bệnh thận do tiểu đường (19).
Các yếu tố tiền viêm và thúc đẩy xơ hoá
Viêm và xơ hóa là những nguyên nhân quan trọng của bệnh thận tiểu đường (20). Cho dù đây là nguyên nhân hay để đáp ứng với tổn thương vẫn còn là một vấn đề tranh luận. Tuy nhiên, có một mối quan hệ chặt chẽ giữa mức độ xâm nhập của đại thực bào và sự xuất hiện sau đó của xơ hóa kẽ ống thận và tiến triển của bệnh thận tiểu đường (21,22).
Đại thực bào bị thu hút vào thận bởi nhiều cơ chế khác nhau (23). Rối loạn chức năng tế bào nội mô, kích hoạt và tổn thương đều kích thích sản xuất các phân tử bám dính trên bề mặt nội mô tạo điều kiện cho sự di chuyển xuyên mô của các đại thực bào. Tổn thương và kích hoạt các tế bào thận cư trú như podocytes (các tế bào có chân), tế bào gian mạch (mesangial cells) và tế bào ống, dẫn đến tiết chemokine tạo điều kiện cho sự xâm nhập của đại thực bào nội thận. Các đại thực bào được kích hoạt theo kiểu hình tiền viêm (M1) bởi ROS, angiotensin II và kích hoạt các thụ thể mineralocorticoid (MR). Điều đó tự nó có thể làm hỏng các tế bào có chân, tế bào nội mô, tế bào gian mạch và tế bào hình ống. Các đại thực bào được kích hoạt, bằng cách giải phóng các cytokine xơ hóa, có thể làm tăng sự tăng sinh tế bào và mở rộng thể tích ma trận và kích thích xơ hóa. Xơ hóa ở cấp độ phân tử được điều tiết một phần do kích hoạt TGFβ1, có hai tác dụng hiệp đồng: kích hoạt yếu tố tăng trưởng mô liên kết (CTGF) và giảm metalloproteinase ma trận (MMPs). Ngược lại, chất đối kháng MR có thể dụ các đại thực bào đến kiểu hình kháng viêm (M2) và được bảo vệ (24). Do đó, đại thực bào đóng một vai trò quan trọng trong sinh bệnh học của bệnh thận tiểu đường (23).
Tổn thương thận cấp tính, viêm, bệnh thận mãn tính và vai trò của MR
Viêm và xơ hóa cũng có thể là những yếu tố thúc đẩy quan trọng sự tiến triển của bệnh thận mãn tính (CKD) ở bệnh nhân tiểu đường, và đây có thể là kết quả của tổn thương thận cấp tính (AKI). Ngày càng có nhiều người nhận ra rằng các đợt AKI đơn lẻ hoặc lặp đi lặp lại trên nền tảng của CKD trong bệnh tiểu đường có thể đóng một vai trò quan trọng trong sự tiến triển của CKD thành ESRD (25). Thâm nhiễm đại thực bào thường thấy ở AKI, và sự suy giảm đại thực bào trong các mô hình tiền lâm sàng có thể bảo vệ khỏi AKI (26). Trong hai mô hình gặm nhấm khác nhau của AKI, điều trị trước tái tưới máu thiếu máu cục bộ hai bên (IR) bằng chất đối kháng mineralocorticoid không steroid finerenone đã ngăn chặn sự phát triển của AKI (27). Trong một loạt các thí nghiệm riêng biệt, tổn thương IR một bên cũng liên quan đến giảm xơ hóa khi động vật được điều trị trước bằng finerenone (27). Hơn nữa, trong một mô hình lợn của IR AKI, việc sử dụng chất đối kháng mineralocorticoid kali canrenoate đã ngăn chặn sự tiến triển của AKI thành CKD sau 90 ngày (27).
Những đóng góp tương đối của việc loại bỏ mineralocorticoid trong các tế bào cơ trơn so với loại trực tiếp của chúng trong các tế bào tủy đã được nghiên cứu trong các mô hình chuột (Hình 4) (27). Với loại bỏ mineralocorticoid trong các tế bào cơ trơn, các mô hình IR đã chứng minh rằng sự gia tăng ngắn hạn của creatinine huyết thanh và nitơ urê máu đã được ngăn chặn. Tuy nhiên, sau 30 ngày, không có sự khác biệt giữa loại trực tiếp MR tế bào cơ hoang dã và tế bào cơ trơn. Trái ngược với việc loại bỏ mineralocorticoid trong các tế bào cơ trơn, trong số những con chuột loại trực tiếp mineralocorticoid dòng tủy, không có sự bảo vệ ngay lập tức khỏi AKI. Tuy nhiên, sau 30 ngày, đã có sự cải thiện rõ rệt về chức năng thận và các dấu hiệu viêm. Hơn nữa, có một sự thay đổi trong sự phân cực của các đại thực bào xâm nhập vào thận. Mặc dù tổng số đại thực bào trong các loại mineralocorticoid hoang dã và loại tủy là tương tự nhau, nhưng đã có sự thay đổi về bản chất của các đại thực bào sao cho các đại thực bào M2 liên quan đến phản ứng kháng viêm đã tăng lên liên quan đến các đại thực bào M1, là tiền viêm (27).
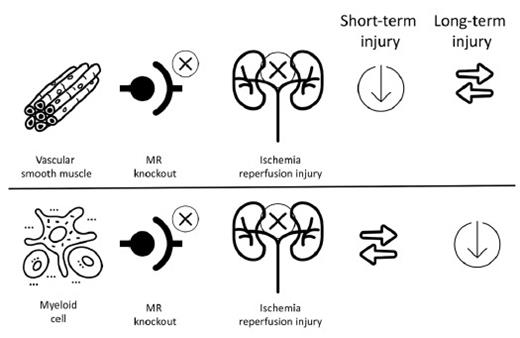
Hình 4. Ảnh hưởng ngắn hạn và dài hạn của MR phụ thuộc vào vị trí. Trong các tế bào cơ trơn, MR bảo vệ khỏi AKI ngắn hạn. Ngược lại, MR trong tế bào tủy không có tác dụng ngắn hạn nhưng ngăn ngừa viêm và xơ hóa lâu dài. Những thí nghiệm này rất hữu ích trong việc tìm hiểu hậu quả lâu dài của AKI lặp đi lặp lại trong sự tiến triển của bệnh thận trong bệnh tiểu đường.
Miễn dịch bẩm sinh, kích hoạt bổ thể và bệnh thận tiểu đường
Kích hoạt hệ thống miễn dịch bẩm sinh thông qua các thụ thể nhận dạng mẫu như thụ thể giống như toll liên kết màng (TLR) và các thụ thể giống như miền oligome hóa liên kết nucleotide (NOD) có thể đóng một vai trò quan trọng trong sinh bệnh học của bệnh thận do tiểu đường (30). Hệ thống bổ thể, ngoài việc chống nhiễm trùng, tạo điều kiện cho việc loại bỏ các tế bào bị tổn thương bởi các kháng thể và tế bào thực bào. Việc kích hoạt thành phần bổ thể C3 tạo ra phức hợp tấn công màng (MAC) làm ly giải, làm hỏng hoặc kích hoạt các tế bào đích. Lectin liên kết Mannose (MBL) kích hoạt con đường lectin; Các phân tử nhận dạng mẫu được gọi là ficolin cũng có thể kích hoạt con đường lectin. Con đường lectin được kích hoạt sau khi liên kết ficolin với protein glycated. Glycation của các protein điều hòa bổ sung như CD59 có thể tự kích hoạt bổ thể; điều này là như vậy bởi vì CD59 thường ức chế MAC (30).
|
Mối quan hệ nhân quả giữa kích hoạt MBL và bệnh thận tiểu đường được thiết lập vững chắc ở động vật. Ví dụ, so với những con chuột hoang dã mắc bệnh tiểu đường do streptozotocin, chuột loại trực tiếp MBL có ít tổn thương thận hơn, ít phì đại thận hơn, bài tiết albumin nước tiểu thấp hơn và biểu hiện collagen loại IV ít hơn (31).
Một số dòng bằng chứng ở người cho thấy vai trò quan trọng của hoạt hóa bổ thể trong tiến triển CKD. Ví dụ, 1) ở bệnh nhân tiểu đường loại 1, nồng độ MBL liên quan đến sự tiến triển của bệnh thận từ macroalbumin niệu sang ESRD (32); 2) trong một nghiên cứu thuần tập tiến cứu trên 270 bệnh nhân mắc bệnh tiểu đường loại 1 mới được chẩn đoán, H-ficolin có liên quan đến việc tăng nguy cơ làm xấu đi albumin niệu (33); và 3) MAC được phát hiện bởi các kháng thể chống lại thành phần C9 của MAC khu trú nó vào màng đáy cầu thận (GBM), ống và bao Bowman ở bệnh nhân tiểu đường loại 1 (34-36).
Kết hợp với nhau, những dữ liệu này chỉ ra vai trò quan trọng của hệ thống bổ thể và các thành phần của nó trong sinh bệnh học của bệnh thận tiểu đường.
Mối tương quan giữa các yếu tố gây bệnh trong bệnh thận tiểu đường
Sự tương tác của các yếu tố chuyển hóa, huyết động, tăng trưởng và xơ hóa được minh họa bằng cách xem xét các thí nghiệm tiền lâm sàng sau đây (37). Các tế bào gian mạch nuôi cấy tiếp xúc với CTGF làm tăng sản xuất các phân tử profibrotic như fibronectin và collagen type I (37). Mặc dù sản xuất CTGF cơ bản bởi các tế bào gian mạch thấp, việc tiếp xúc của các tế bào gian mạch với nồng độ glucose tăng (một yếu tố trao đổi chất) hoặc căng thẳng chuyển hóa theo chu kỳ (một yếu tố huyết động) làm tăng sản xuất CTGF (một yếu tố tăng trưởng). Sự cảm ứng protein CTGF bởi nồng độ glucose cao bị chặn bởi kháng thể trung hòa TGFβ1. Điều này cho thấy rằng một yếu tố tăng trưởng khác – TGFβ1 – làm trung gian ảnh hưởng của nồng độ glucose cao để kích thích sản xuất CTGF. Các nghiên cứu in vivo ở chuột mắc bệnh tiểu đường db / db béo phì chứng minh rằng phiên mã CTGF đã tăng gấp 28 lần sau ~ 3,5 tháng mắc bệnh tiểu đường (37). Khi mắc bệnh tiểu đường 3,5 tháng, sự giãn nở tế bào gian mạch là nhẹ, và không có bệnh kẽ và protein niệu. Hơn nữa, thay vì tăng khuếch tán khắp thận, việc sản xuất CTGF chỉ giới hạn ở khoang cầu thận. Những thí nghiệm này chứng minh sự tương tác của tất cả các yếu tố gây bệnh được thảo luận ở trên và nhấn mạnh mối tương quan phức tạp của các yếu tố này, theo thời gian và tại các vị trí khác nhau trong thận, trong việc tạo ra các biểu hiện mô học của bệnh thận tiểu đường.
Phân loại giải phẫu bệnh học của bệnh thận tiểu đường
Theo một hội nghị đồng thuận quốc tế, các biểu hiện mô học của bệnh thận đái tháo đường tuân theo bốn lớp tiến triển (Bảng 2) (38). Việc phân loại thừa nhận các tổn thương ở cầu thận, ống thận và mạch máu, nhưng gốc của hệ thống phân loại dựa trên sự xuất hiện của cầu thận. Theo hệ thống phân loại này, bệnh thận do tiểu đường tiến triển từ dày GBM, đến giãn nở gian mạch, tổn thương Kimmelstiel-Wilson và xơ cứng cầu thận lan toả, được phản ánh trong bốn lớp, như được thảo luận thêm dưới đây. Mặc dù hệ thống này chưa được xác nhận với kết quả lâm sàng, nhưng nó đóng vai trò là một công cụ nghiên cứu và lâm sàng quan trọng để phân loại mức độ nghiêm trọng của tổn thương thận do tiểu đường.
| Lớp I | Màng đáy cầu thận dày lên trên kính hiển vi điện tử; Tối thiểu, không đặc hiệu hoặc không có thay đổi trên kính hiển vi quang học |
| Lớp II |
|
| Lớp IIa |
|
| Lớp IIb |
|
| Lớp III |
|
| Lớp IV |
|
Kết luận
Cơ chế bệnh sinh của bệnh thận tiểu đường tương tự ở bệnh tiểu đường loại 1 và loại 2. Bệnh thận tiểu đường được phân loại mô học bằng sự biểu hiện của cầu thận trên sinh thiết thận. Nó tiến triển từ sự dày lên của màng đáy cầu thận, đến giãn nở gian mạch, xơ cứng cầu thận dạng nốt và xơ cứng cầu thận lan toả. Cầu thận to, tổn thương mạch máu, IFTA và các giọt tái hấp thu ở ống thận đều thường gặp. Cơ chế bệnh sinh của bệnh thận đái tháo đường liên quan đến các yếu tố chuyển hóa, huyết động, tăng trưởng và các yếu tố viêm và xơ hóa. Sự đóng góp tương đối của các yếu tố này khác nhau giữa các bệnh nhân, theo thời gian và thậm chí ở các khoang khác nhau của thận, và các yếu tố di truyền và môi trường có thể thay đổi sự xuất hiện của các tổn thương thận. AKI đóng một vai trò quan trọng trong sự tiến triển của bệnh thận ở bệnh nhân tiểu đường. Kích hoạt thụ thể mineralocorticoid, đặc biệt là trong các tế bào tủy, có thể quan trọng trong việc làm trung gian viêm và xơ hóa trong CKD và sau AKI ở những người mắc bệnh tiểu đường loại 2, và liệu pháp đối kháng thụ thể mineralocorticoid có thể bảo vệ.
TRÍCH DẪN TÀI LIỆU GỐC:
Rajiv Agarwal; Pathogenesis of Diabetic Nephropathy. ADA Clinical Compendia 1 June 2021; 2021 (1): 2–7. https://doi.org/10.2337/db20211-2
TÀI LIỆU THAM KHẢO
1.Ali MK, Bullard KM, Saydah S, Imperatore G, Gregg EW. Cardiovascular and renal burdens of prediabetes in the USA: analysis of data from serial cross-sectional surveys, 1988–2014. Lancet Diabetes Endocrinol 2018;6:392–403
2.Afkarian M, Sachs MC, Kestenbaum B, et al Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol 2013;24:302–308
3.Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861–869
4.Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345: 851–860
5.Wanner C, Inzucchi SE, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016;375:323–334
6.Heerspink HJL, Desai M, Jardine M, Balis D, Meininger G, Perkovic V. Canagliflozin slows progression of renal function decline independently of glycemic effects. J Am Soc Nephrol 2017;28:368–375
7.Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016;375:1834–1844
8.Perkovic V, Jardine MJ, Neal B, et al; CREDENCE Trial Investigators. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 2019;380:2295–2306
9.Bakris GL, Agarwal R, Anker SD, et al; FIDELIO-DKD Investigators. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med 2020;383:2219–2229
10.White KE, Bilous RW. Type 2 diabetic patients with nephropathy show structural-functional relationships that are similar to type 1 disease. J Am Soc Nephrol 2000;11:1667–1673
11.Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 2002;288:2579–2588
12.Vallon V, Komers R. Pathophysiology of the diabetic kidney. Compr Physiol 2011;1:1175–1232
13.Sorbinil Retinopathy Trial Research Group. A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Arch Ophthalmol 1990;108:1234–1244
14.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl 2000;77:S13–S18
15.Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 2005;28: 2686–2690
16.Tonneijck L, Muskiet MH, Smits MM, et al Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol 2017;28:1023–1039
17.Brenner BM, Lawler EV, Mackenzie HS. The hyperfiltration theory: a paradigm shift in nephrology. Kidney Int 1996;49:1774–1777
18.Cooper ME, Vranes D, Youssef S, et al Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999;48:2229–2239
19.Gnudi L. Angiopoietins and diabetic nephropathy. Diabetologia 2016;59: 1616–1620
20.Bohle A, Wehrmann M, Bogenschutz O, Batz C, Müller CA, Müller GA. The pathogenesis of chronic renal failure in diabetic nephropathy: investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract 1991;187:251–259
21.Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006;11:226–231
22.Yonemoto S, Machiguchi T, Nomura K, Minakata T, Nanno M, Yoshida H. Correlations of tissue macrophages and cytoskeletal protein expression with renal fibrosis in patients with diabetes mellitus. Clin Exp Nephrol 2006;10:186–192
23.Tesch GH. Macrophages and diabetic nephropathy. Semin Nephrol 2010;30:290–301
24.Bertocchio JP, Warnock DG, Jaisser F. Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. Kidney Int 2011;79:1051–1060
25.Yu SM, Bonventre JV. Acute kidney injury and progression of diabetic kidney disease. Adv Chronic Kidney Dis 2018;25:166–180
26.Kinsey GR. Macrophage dynamics in AKI to CKD progression. J Am Soc Nephrol 2014;25:209–211
27.Barrera-Chimal J, Estrela GR, Lechner SM, et al The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int 2018;93:1344–1355
28.Wang Y, Harris DC. Macrophages in renal disease. J Am Soc Nephrol 2011; 22:21–27
29.Calle P, Hotter G. Macrophage phenotype and fibrosis in diabetic nephropathy. Int J Mol Sci 2020;21:2806
30.Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol 2017;13:311–318
31.Østergaard J, Thiel S, Gadjeva M, Hansen TK, Rasch R, Flyvbjerg A. Mannose-binding lectin deficiency attenuates renal changes in a streptozotocin-induced model of type 1 diabetes in mice. Diabetologia 2007;50:1541–1549
32.Hansen TK, Forsblom C, Saraheimo M, et al Association between mannose-binding lectin, high-sensitivity C-reactive protein and the progression of diabetic nephropathy in type 1 diabetes. Diabetologia 2010;53:1517–1524
33.Østergaard JA, Thiel S, Hovind P, et al Association of the pattern recognition molecule H-ficolin with incident microalbuminuria in an inception cohort of newly diagnosed type 1 diabetic patients: an 18 year follow-up study. Diabetologia 2014;57:2201–2207
34.Falk RJ, Dalmasso AP, Kim Y, et al Neoantigen of the polymerized ninth component of complement: characterization of a monoclonal antibody and immunohistochemical localization in renal disease. J Clin Invest 1983;72:560–573
35.Falk RJ, Scheinman JI, Mauer SM, Michael AF. Polyantigenic expansion of basement membrane constituents in diabetic nephropathy. Diabetes 1983;32(Suppl. 2):34–39
36.Falk RJ, Sisson SP, Dalmasso AP, Kim Y, Michael AF, Vernier RL. Ultrastructural localization of the membrane attack complex of complement in human renal tissues. Am J Kidney Dis 1987;9:121–128
37.Riser BL, Denichilo M, Cortes P, et al Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol 2000;11:25–38
38.Tervaert TW, Mooyaart AL, Amann K, et al Pathologic classification of diabetic nephropathy. J Am Soc Nephrol 2010;21:556–563
BÌNH LUẬN