
Phác đồ chẩn đoán và điều trị Hội chứng Fanconi
THƯ VIỆN MEDIPHARM
1. Đại cương
1.1. Định nghĩa
Hội chứng Fanconi là một rối loạn chức năng tổng quát của ống lượn gần (PCT) dẫn đến tăng bài tiết nhiều chất như amino acid, glucose, phosphate, bicarbonate, kali, acid uric, protein trọng lượng phân tử thấp và các chất hòa tan khác.
1.2. Dịch tễ học
- Tỷ lệ mắc: Hiếm gặp, chưa có số liệu chính xác.
- Hội chứng Fanconi di truyền được báo cáo xảy ra khoảng 1/40.000 trẻ sinh ra.
- Hội chứng Fanconi di truyền thường gặp ở trẻ em hơn người lớn.
- Hội chứng Fanconi mắc phải thường gặp ở người lớn hơn trẻ em, nhưng có thể xảy ra ở mọi lứa tuổi.
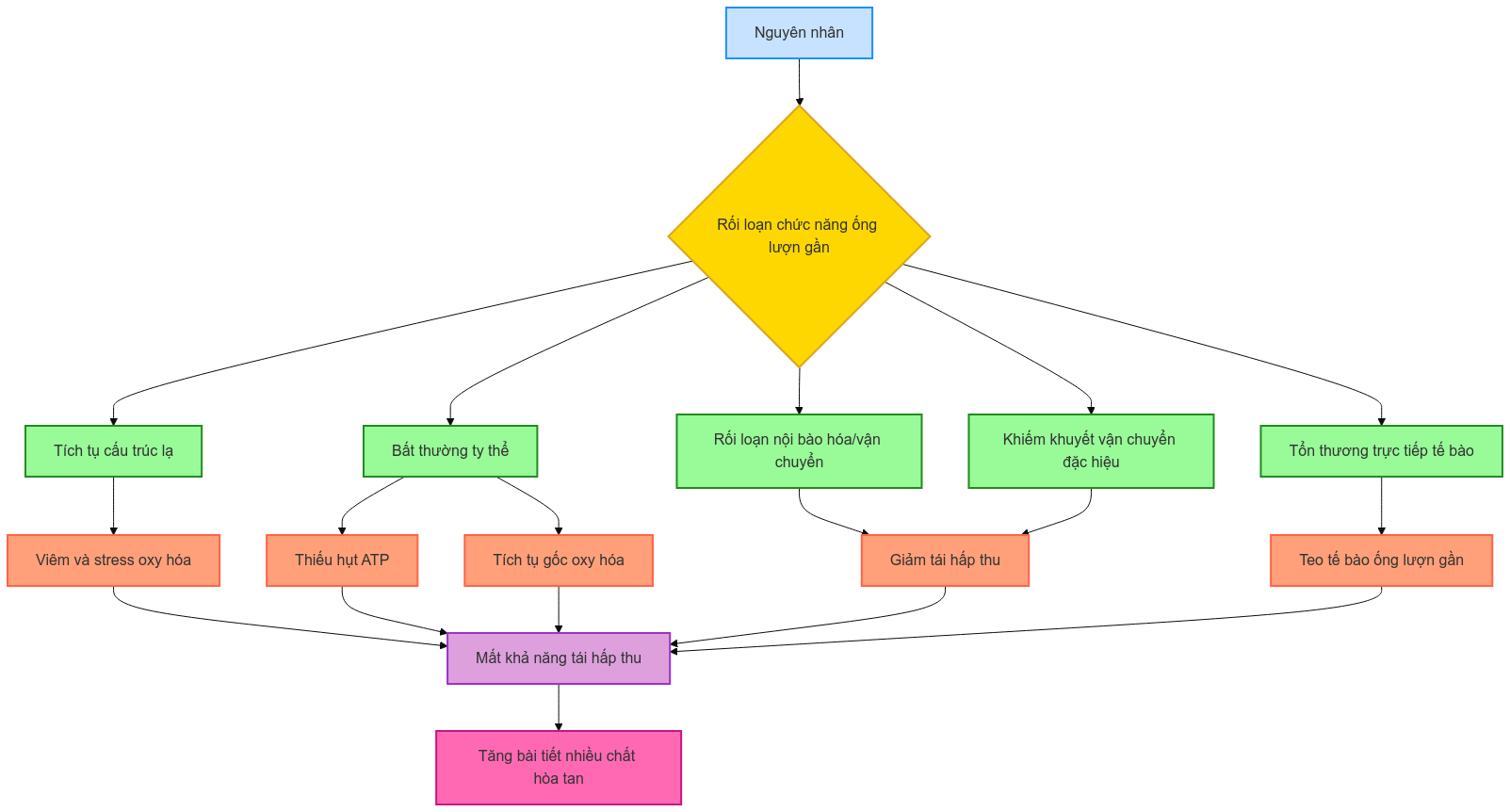
1.3. Sinh lý bệnh
Hội chứng Fanconi xảy ra do rối loạn chức năng tái hấp thu của ống lượn gần. Cơ chế bệnh sinh chính xác chưa được hiểu rõ hoàn toàn và có thể khác nhau tùy theo nguyên nhân, nhưng có thể bao gồm:
- Tích tụ cấu trúc lạ:
- Ví dụ: Tinh thể cystine trong bệnh cystin
- Chuỗi nhẹ immunoglobulin trong bệnh đa u tủy xương
- Bất thường ty thể:
- Gián đoạn chuỗi vận chuyển electron
- Suy giảm phosphoryl hóa oxy hóa
- Dẫn đến thiếu hụt ATP và tích tụ các gốc oxy hóa
- Rối loạn nội bào hóa và vận chuyển nội bào:
- Ví dụ: Bất thường cytoskeleton trong hội chứng Lowe và bệnh Dent type 2
- Khiếm khuyết vận chuyển đặc hiệu:
- Ví dụ: Khiếm khuyết protein vận chuyển glucose GLUT2 trong hội chứng Fanconi-Bickel
- Tổn thương trực tiếp tế bào ống lượn gần:
- Do cơ chế tự miễn
- Do độc tính thuốc
- Do đáp ứng nội bào với sự suy giảm phiên mã
Tóm tắt cơ chế sinh lý bệnh hội chứng Fanconi
1.4. Nguyên nhân
Hội chứng Fanconi có thể do di truyền hoặc mắc phải:
- Hội chứng Fanconi di truyền:
- Nguyên phát: Do đột biến gen GATM, SLC34A1, EHHADH, HNF4A, NDUFAF6
- Thứ phát: Do các bệnh di truyền hệ thống như cystinosis, bệnh Dent, hội chứng Lowe, bệnh Wilson, v.v.
- Hội chứng Fanconi mắc phải:
- Do thuốc: Tenofovir, gentamicin, ifosfamide, valproic acid, v.v.
- Do bệnh tự miễn: Hội chứng Sjögren, viêm thận-bể thận kẽ và viêm màng bồ đào (TINU)
- Do bệnh tương bào: Đa u tủy xương, bệnh chuỗi nhẹ đơn dòng
- Do nhiễm độc kim loại nặng: Chì, cadmium, thủy ngân
- Do độc chất: Paraquat, toluene
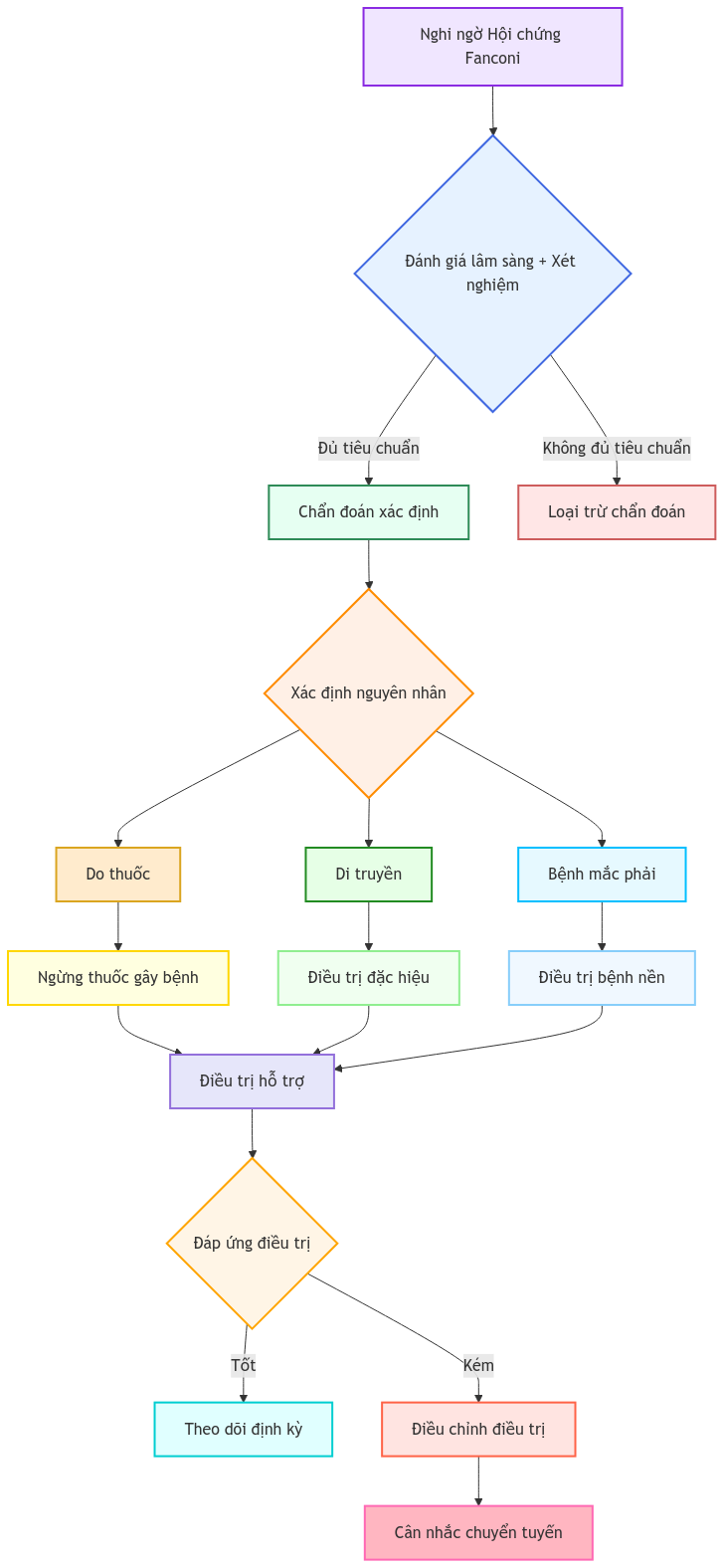
2. Chẩn đoán
2.1. Lâm sàng
- Triệu chứng có thể khác nhau tùy theo nguyên nhân và mức độ tổn thương ống lượn gần.
- Các triệu chứng thường gặp:
- Đa niệu, khát nước
- Mệt mỏi, suy nhược
- Đau xương, gãy xương bệnh lý
- Yếu cơ
- Chậm tăng trưởng (ở trẻ em)
2.2. Cận lâm sàng
2.2.1. Xét nghiệm máu
- Điện giải đồ: Hạ kali, hạ phosphate, hạ natri, hạ calci, hạ magiê
- Khí máu: Toan chuyển hóa (bicarbonate thấp)
- Acid uric máu thấp
- Glucose máu bình thường
- Creatinine có thể tăng
2.2.2. Xét nghiệm nước tiểu
- Đường niệu (khi glucose máu bình thường)
- Aminoacid niệu
- Protein niệu trọng lượng phân tử thấp (như β2-microglobulin)
- Phosphate niệu tăng
- Calci niệu tăng
- Acid uric niệu tăng
- pH nước tiểu > 5.5 khi có toan chuyển hóa
2.2.3. Xét nghiệm chuyên sâu
- Đo bài tiết phân đoạn các chất trong nước tiểu 24 giờ
- Sinh thiết thận (trong một số trường hợp)
- Xét nghiệm di truyền (nếu nghi ngờ nguyên nhân di truyền)
2.3. Tiêu chuẩn chẩn đoán
Chẩn đoán xác định khi có ít nhất 3 trong 5 tiêu chuẩn sau:
- Đường niệu khi glucose máu bình thường
- Aminoacid niệu toàn phần
- Phosphate niệu tăng (FEPi > 5% hoặc TmP/GFR giảm)
- Acid uric niệu tăng (FEUA > 10%)
- Protein niệu trọng lượng phân tử thấp (β2-microglobulin niệu > 0.3 mg/L)
2.4. Chẩn đoán phân biệt
| Bệnh | Điểm giống | Điểm khác biệt | Cách phân biệt |
|---|---|---|---|
| Toan ống thận xa (RTA type 1) | Toan chuyển hóa | pH nước tiểu > 5.5 khi có toan | Không có các rối loạn tái hấp thu khác của ống lượn gần |
| Đái tháo đường | Đa niệu, khát nước | Glucose máu tăng | Glucose máu bình thường trong hội chứng Fanconi |
| Hội chứng Bartter | Hạ kali máu | Kiềm chuyển hóa | Toan chuyển hóa trong hội chứng Fanconi |
| Cường cận giáp | Hạ phosphate máu | Calci máu tăng | Calci máu bình thường hoặc thấp trong hội chứng Fanconi |
3. Điều trị
3.1. Nguyên tắc điều trị
- Điều trị nguyên nhân (nếu có thể)
- Bù đắp các chất điện giải và acid-base bị mất
- Điều trị các biến chứng
- Theo dõi và quản lý lâu dài
3.2. Điều trị cụ thể
3.2.1. Điều trị nguyên nhân
- Ngừng thuốc gây độc thận (nếu nguyên nhân do thuốc)
- Điều trị bệnh nền (ví dụ: điều trị đa u tủy xương, hội chứng Sjögren)
- Điều trị đặc hiệu cho các bệnh di truyền (ví dụ: cysteamine cho bệnh cystinosis)
3.2.2. Điều trị hỗ trợ
a. Bù nước và điện giải:
- Natri: 1-3 mEq/kg/ngày, dạng NaCl hoặc NaHCO3
- Kali: 2-10 mEq/kg/ngày, dạng KCl hoặc Kali citrate
- Phosphate: 30-60 mg/kg/ngày phosphate trung tính
- Calci: 30-75 mg/kg/ngày calci carbonate
- Magiê: 10-20 mg/kg/ngày Mg oxide hoặc Mg gluconate
b. Điều chỉnh toan chuyển hóa:
- Natri bicarbonate: 1-2 mEq/kg/ngày, chia 3-4 lần
- Mục tiêu: duy trì bicarbonate máu > 22 mEq/L
c. Vitamin D và các chất bổ sung khác:
- Calcitriol: 20-60 ng/kg/ngày
- Carnitine: 50-100 mg/kg/ngày (nếu thiếu)
d. Điều trị còi xương/loãng xương:
- Kết hợp bổ sung calci, phosphate và vitamin D
- Có thể cần thuốc chống hủy xương trong một số trường hợp
3.3. Theo dõi và đánh giá
- Đánh giá lâm sàng: 1-3 tháng/lần
- Xét nghiệm máu (điện giải, chức năng thận, khí máu): 1-3 tháng/lần
- Xét nghiệm nước tiểu 24 giờ: 3-6 tháng/lần
- Đánh giá tăng trưởng và phát triển ở trẻ em: 3-6 tháng/lần
- Đánh giá mật độ xương: 1-2 năm/lần
3.4. Tiêu chí chuyển tuyến/hội chẩn chuyên khoa
- Khó kiểm soát rối loạn điện giải hoặc acid-base
- Suy giảm chức năng thận tiến triển
- Biến chứng xương nặng
4. Tiên lượng
Tiên lượng của hội chứng Fanconi phụ thuộc vào nguyên nhân cơ bản và mức độ nghiêm trọng của bệnh:
- Hội chứng Fanconi mắc phải do thuốc: Thường cải thiện sau khi ngừng thuốc gây bệnh, nhưng có thể mất vài tháng và không phải lúc nào cũng hồi phục hoàn toàn.
- Mức độ bằng chứng: Trung bình, Khuyến nghị: Mạnh
- Hội chứng Fanconi di truyền: Tiên lượng kém hơn, thường cần điều trị suốt đời.
- Trong bệnh cystinosis không điều trị, suy thận thường xảy ra vào khoảng 6-12 tuổi.
- Điều trị bằng cysteamine có thể trì hoãn sự tiến triển đến suy thận nhưng không đảo ngược hội chứng Fanconi.
- Mức độ bằng chứng: Cao, Khuyến nghị: Mạnh
- Hội chứng Fanconi liên quan đến bệnh đa u tủy xương: Thường tiến triển đến bệnh thận giai đoạn cuối.
- Mức độ bằng chứng: Trung bình, Khuyến nghị: Mạnh
- Biến chứng lâu dài có thể bao gồm:
- Suy thận mạn tính
- Còi xương hoặc loãng xương
- Chậm tăng trưởng ở trẻ em
- Rối loạn điện giải mạn tính
5. Phòng bệnh
5.1. Phòng bệnh tiên phát
- Tư vấn di truyền cho các gia đình có tiền sử mắc hội chứng Fanconi di truyền.
- Tránh phơi nhiễm với các kim loại nặng và độc chất công nghiệp.
5.2. Phòng bệnh thứ phát
- Đối với bệnh nhân đang sử dụng các thuốc có nguy cơ gây độc thận:
- Theo dõi chặt chẽ chức năng ống lượn gần.
- Sử dụng liều thuốc tối thiểu cần thiết để đạt hiệu quả điều trị.
- Điều chỉnh liều thuốc theo chức năng thận.
- Mức độ bằng chứng: Trung bình, Khuyến nghị: Mạnh
- Đối với bệnh nhân HIV đang điều trị bằng thuốc kháng retrovirus:
- Theo dõi định kỳ điện giải đồ và độ thanh thải creatinine.
- Tính toán độ thanh thải creatinine trước khi bắt đầu điều trị, ngay cả khi creatinine huyết thanh bình thường.
- Mức độ bằng chứng: Cao, Khuyến nghị: Mạnh
6. Hướng dẫn cho bệnh nhân
- Giáo dục bệnh nhân và gia đình về bản chất của bệnh và tầm quan trọng của việc tuân thủ điều trị.
- Hướng dẫn cách nhận biết các dấu hiệu cảnh báo cần đến cơ sở y tế:
- Mệt mỏi tăng
- Yếu cơ
- Đau xương nghiêm trọng
- Giảm lượng nước tiểu
- Tư vấn về chế độ ăn:
- Đảm bảo đủ nước và điện giải
- Chế độ ăn giàu kali, phosphate (nếu không có chống chỉ định)
- Hạn chế natri nếu có tăng huyết áp
- Hướng dẫn về cách sử dụng thuốc và thực phẩm bổ sung:
- Cách uống thuốc đúng liều và đúng thời điểm
- Tầm quan trọng của việc không bỏ liều
- Khuyến khích tập thể dục phù hợp để duy trì sức khỏe xương.
- Đối với trẻ em, cần theo dõi sát sự tăng trưởng và phát triển, báo cáo bất kỳ lo ngại nào với bác sĩ.
- Lập kế hoạch theo dõi và tái khám định kỳ.
Tài liệu tham khảo
- Bökenkamp A, Ludwig M. Disorders of the renal proximal tubule. Nephron Physiol. 2011;118(1):1-6.
- Kleta R, Gahl WA. Pharmacological treatment of nephropathic cystinosis with cysteamine. Expert Opin Pharmacother. 2004;5(11):2255-2262.
- Hall AM, Bass P, Unwin RJ. Drug-induced renal Fanconi syndrome. QJM. 2014;107(4):261-269.
- Izzedine H, Launay-Vacher V, Isnard-Bagnis C, Deray G. Drug-induced Fanconi’s syndrome. Am J Kidney Dis. 2003;41(2):292-309.
- Foreman JW. Fanconi Syndrome. Pediatr Clin North Am. 2019;66(1):159-167.
- Albuquerque ALB, Dos Santos Borges R, Conegundes AF, et al. Inherited Fanconi syndrome. World J Pediatr. 2023;19(7):619-634.
- Zandi-Nejad K, Eddy AA, Glassock RJ, Brenner BM. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int Suppl. 2004;(92):S76-S89.
- Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant. 2012;27(12):4273-4287.
BÌNH LUẬN