
Phác đồ chẩn đoán và điều trị Hội chứng Crigler-Najjar
THƯ VIỆN MEDIPHARM
1. Đại cương
1.1. Định nghĩa
Hội chứng Crigler-Najjar (CN) là một bệnh di truyền hiếm gặp, đặc trưng bởi tăng bilirubin gián tiếp (không liên hợp) trong máu do thiếu hụt hoàn toàn hoặc một phần enzyme UDP-glucuronosyltransferase 1A1 (UGT1A1) ở gan.
Tên gọi “Hội chứng Crigler-Najjar” có nguồn gốc từ tên của hai bác sĩ người Mỹ đã mô tả đầu tiên về hội chứng này vào năm 1952. Cụ thể:
- John Fielding Crigler Jr. (1919-2018): Là một bác sĩ nhi khoa nổi tiếng tại Bệnh viện Nhi Boston và Trường Y Harvard. Ông có nhiều đóng góp quan trọng trong lĩnh vực nhi khoa và nội tiết học nhi khoa.
- Victor Assad Najjar (1914-2002): Là một bác sĩ và nhà nghiên cứu y học tại Đại học Johns Hopkins. Ông có nhiều công trình nghiên cứu về các bệnh di truyền ở trẻ em.
Vào năm 1952, Crigler và Najjar đã công bố một báo cáo về 7 trường hợp trẻ sơ sinh trong cùng một gia đình bị vàng da nặng và tử vong sớm do tăng bilirubin máu nghiêm trọng. Họ đã mô tả chi tiết về các triệu chứng, diễn biến lâm sàng và đặc điểm di truyền của bệnh này.
Báo cáo của họ đã được xuất bản trên tạp chí Pediatrics với tiêu đề “Congenital familial nonhemolytic jaundice with kernicterus” (Vàng da bẩm sinh gia đình không tan máu kèm vàng da nhân). Đây là mô tả đầu tiên về hội chứng này trong y văn.
Sau đó, cộng đồng y học đã đặt tên cho hội chứng này là “Hội chứng Crigler-Najjar” để vinh danh công trình nghiên cứu tiên phong của hai bác sĩ này. Tên gọi này đã được sử dụng rộng rãi trong y văn và thực hành lâm sàng cho đến ngày nay.
1.2. Dịch tễ học
- Tỷ lệ mắc: Khoảng 1 trên 1,000,000 trẻ sinh ra.
- Phân bố: Gặp ở cả nam và nữ, không phụ thuộc chủng tộc.
- Di truyền: Theo kiểu lặn trên nhiễm sắc thể thường.
1.3. Sinh lý bệnh
- Nguyên nhân: Đột biến gen UGT1A1 trên nhiễm sắc thể 2q37.1.
- Cơ chế:
- Thiếu hụt enzyme UGT1A1 dẫn đến giảm khả năng liên hợp bilirubin.
- Bilirubin không liên hợp tích tụ trong máu.
- Khi nồng độ cao, bilirubin có thể đi qua hàng rào máu não gây độc cho não.
Lược đồ cơ chế sinh lý bệnh:

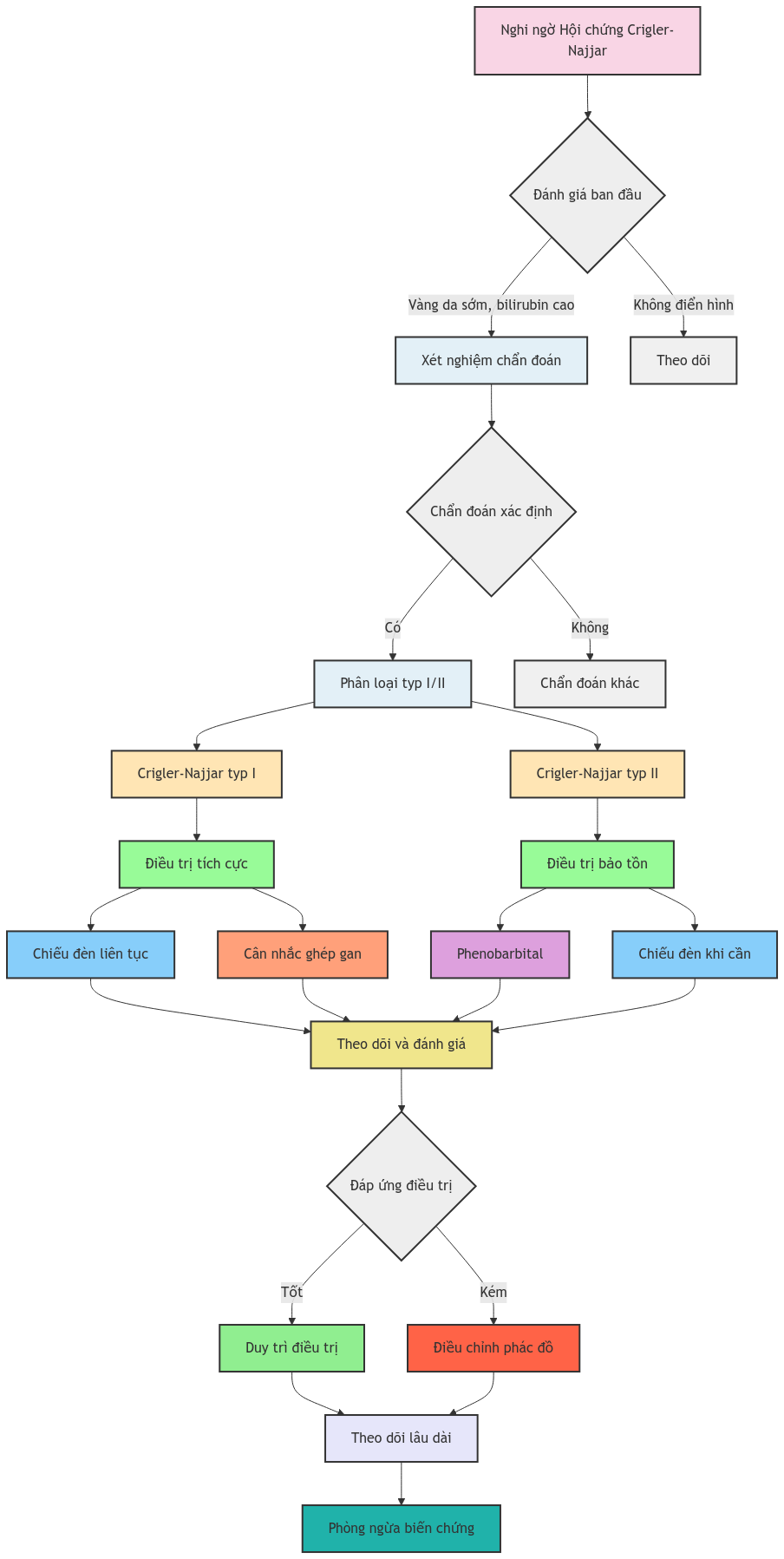
1.4. Phân loại
- Hội chứng Crigler-Najjar typ I (CN-I):
- Thiếu hụt hoàn toàn enzyme UGT1A1.
- Nồng độ bilirubin không liên hợp rất cao (thường > 20 mg/dL).
- Không đáp ứng với điều trị phenobarbital.
- Hội chứng Crigler-Najjar typ II (CN-II):
- Thiếu hụt một phần enzyme UGT1A1 (< 10% hoạt tính bình thường).
- Nồng độ bilirubin không liên hợp thấp hơn (thường < 20 mg/dL).
- Đáp ứng với điều trị phenobarbital.
2. Chẩn đoán
2.1. Lâm sàng
- Vàng da sớm sau sinh (trong 24-48 giờ đầu đối với CN-I).
- Không có dấu hiệu tan máu hoặc bệnh lý gan.
- Có thể có biểu hiện thần kinh (đặc biệt trong CN-I) như:
- Kích thích
- Co giật
- Opisthotonos (ưỡn cong lưng)
- Giảm trương lực cơ
2.2. Cận lâm sàng
2.2.1. Xét nghiệm máu
- Bilirubin toàn phần và bilirubin trực tiếp:
- CN-I: Bilirubin gián tiếp > 20 mg/dL (342 μmol/L)
- CN-II: Bilirubin gián tiếp thường < 20 mg/dL
- Công thức máu: Bình thường
- Chức năng gan: Bình thường
- Albumin máu: Bình thường hoặc giảm nhẹ
2.2.2. Xét nghiệm di truyền
- Phân tích đột biến gen UGT1A1
- Xét nghiệm hoạt tính enzyme UGT1A1 (nếu có thể)
2.2.3. Xét nghiệm khác
- Phân tích mẫu phân hoặc dịch dạ dày để tìm bilirubin liên hợp (có thể hiện diện trong CN-II)
- Siêu âm gan: Loại trừ các bất thường cấu trúc
2.3. Tiêu chuẩn chẩn đoán
- Tăng bilirubin gián tiếp mà không có bằng chứng của tan máu hoặc bệnh gan.
- Bilirubin gián tiếp ≥ 20 mg/dL (342 μmol/L) đối với CN-I.
- Xác định đột biến gen UGT1A1.
- Đáp ứng với phenobarbital (giảm bilirubin > 30% sau 2-3 ngày) gợi ý CN-II.
2.4. Chẩn đoán phân biệt
| Bệnh | Điểm giống | Điểm khác biệt | Cách phân biệt |
|---|---|---|---|
| Vàng da sinh lý ở trẻ sơ sinh | Vàng da sớm | Bilirubin thường < 15 mg/dL, tự giới hạn | Theo dõi diễn biến, xét nghiệm bilirubin |
| Tăng bilirubin do tan máu | Vàng da, tăng bilirubin gián tiếp | Có dấu hiệu tan máu, thiếu máu | Công thức máu, test Coombs |
| Hội chứng Gilbert | Tăng bilirubin gián tiếp | Mức độ nhẹ hơn, thường không có triệu chứng | Xét nghiệm di truyền, đáp ứng với nhịn đói |
| Bệnh gan bẩm sinh khác | Vàng da kéo dài | Có thể có tăng men gan, hepatomegaly | Siêu âm gan, sinh thiết gan nếu cần |
2.5. Đánh giá
- Đánh giá mức độ tăng bilirubin và nguy cơ nhiễm độc thần kinh.
- Đánh giá phát triển thần kinh và tâm thần vận động.
- Đánh giá chức năng gan và các cơ quan khác.
- Đánh giá khả năng tuân thủ điều trị lâu dài.
3. Điều trị
3.1. Nguyên tắc điều trị
- Giảm nồng độ bilirubin không liên hợp trong máu
- Ngăn ngừa biến chứng thần kinh
- Điều trị hỗ trợ và theo dõi lâu dài
3.2. Điều trị cụ thể
3.2.1. Điều trị không dùng thuốc
a. Chiếu đèn (Phototherapy):
- CN-I: Chiếu đèn liên tục hoặc 12-16 giờ/ngày.
- CN-II: Chiếu đèn khi cần thiết.
- Cường độ: 30 μW/cm²/nm hoặc cao hơn.
- Bước sóng: 460-490 nm (ánh sáng xanh).
b. Chế độ dinh dưỡng:
- Khuyến khích bú mẹ.
- Tránh nhịn đói kéo dài.
- Bổ sung calori và protein đầy đủ.
3.2.2. Điều trị dùng thuốc
a. Phenobarbital:
- Liều: 4-5 mg/kg/ngày chia 2 lần.
- Chỉ định: CN-II và một số trường hợp CN-I có đáp ứng.
- Thời gian: Có thể dùng lâu dài nếu hiệu quả.
- Theo dõi: Chức năng gan, tác dụng an thần.
b. Calcium phosphate hoặc Cholestyramine:
- Liều: Tùy theo đáp ứng lâm sàng.
- Cơ chế: Giúp giảm tái hấp thu bilirubin từ ruột.
- Theo dõi: Cân bằng điện giải, hấp thu vitamin tan trong mỡ.
c. Tin-mesoporphyrin (Sn-MP):
- Liều: 6 μmol/kg, tiêm bắp mỗi 7-10 ngày.
- Cơ chế: Ức chế heme oxygenase, giảm sản xuất bilirubin.
- Lưu ý: Chưa được FDA phê duyệt, sử dụng trong nghiên cứu.
3.2.3. Điều trị can thiệp
a. Thay máu (Exchange transfusion):
- Chỉ định: Khi bilirubin > 20-25 mg/dL ở trẻ sơ sinh hoặc khi có dấu hiệu nhiễm độc thần kinh.
- Kỹ thuật: Thay thế 2 lần thể tích máu của trẻ.
- Theo dõi: Cân bằng điện giải, đông máu, nhiễm trùng.
b. Ghép gan:
- Chỉ định: CN-I không đáp ứng với các biện pháp điều trị khác.
- Thời điểm: Nên cân nhắc sớm, trước khi có tổn thương thần kinh không hồi phục.
- Kết quả: Tỷ lệ sống sau 1 năm > 90%.
3.3. Điều trị theo giai đoạn
a. Sơ sinh:
- Chiếu đèn tích cực.
- Theo dõi bilirubin chặt chẽ mỗi 4-6 giờ.
- Thay máu nếu cần.
b. Trẻ nhỏ và trẻ lớn:
- CN-I: Chiếu đèn liên tục, cân nhắc ghép gan.
- CN-II: Phenobarbital, chiếu đèn khi cần.
c. Người lớn:
- Tiếp tục điều trị duy trì.
- Theo dõi biến chứng lâu dài.
3.4. Điều trị dự phòng
- Tránh các yếu tố làm tăng bilirubin: nhiễm trùng, nhịn đói, thuốc gây tan máu.
- Tiêm phòng viêm gan B.
- Tư vấn di truyền cho gia đình.
4. Theo dõi và đánh giá
4.1. Theo dõi
- Định kỳ xét nghiệm bilirubin máu:
- Hàng ngày trong tuần đầu.
- Hàng tuần trong 3 tháng đầu.
- Hàng tháng cho đến 1 tuổi.
- Sau đó 3-6 tháng/lần.
- Đánh giá phát triển thần kinh định kỳ:
- Mỗi 3-6 tháng trong 2 năm đầu.
- Hàng năm sau đó.
- Kiểm tra thính lực và thị lực hàng năm.
- Theo dõi tác dụng phụ của thuốc (nếu có sử dụng).
- Siêu âm gan hàng năm để theo dõi sỏi mật.
4.2. Tiêu chí đánh giá hiệu quả điều trị
- Duy trì nồng độ bilirubin dưới ngưỡng gây độc thần kinh:
- < 20 mg/dL (342 μmol/L) đối với CN-I.
- < 15 mg/dL (257 μmol/L) đối với CN-II.
- Không có biểu hiện nhiễm độc thần kinh cấp hoặc mạn tính.
- Phát triển thể chất và tâm thần vận động bình thường.
- Chất lượng cuộc sống được cải thiện.
5. Phòng bệnh
5.1. Phòng bệnh tiên phát
- Tư vấn di truyền cho các cặp vợ chồng có nguy cơ.
- Chẩn đoán trước sinh nếu có tiền sử gia đình:
- Phân tích ADN từ tế bào ối hoặc gai nhau.
- Thực hiện từ tuần 11-13 của thai kỳ.
5.2. Phòng bệnh thứ phát
- Giáo dục bệnh nhân và gia đình về:
- Tầm quan trọng của việc tuân thủ điều trị.
- Nhận biết các dấu hiệu cảnh báo của tăng bilirubin.
- Tránh các yếu tố làm tăng bilirubin.
- Tiêm phòng đầy đủ, đặc biệt là vaccine viêm gan B.
- Kiểm soát chặt chẽ các bệnh lý kèm theo.
6. Tiên lượng
6.1. Crigler-Najjar typ I
- Tiên lượng nặng nếu không được điều trị tích cực.
- Nguy cơ cao bị kernicterus và tử vong trong những năm đầu đời.
- Ghép gan có thể cải thiện tiên lượng đáng kể:
- Tỷ lệ sống sau 1 năm > 90%.
- Chất lượng cuộc sống được cải thiện.
6.2. Crigler-Najjar typ II
- Tiên lượng tốt hơn so với typ I.
- Đa số bệnh nhân có thể sống đến tuổi trưởng thành với chất lượng cuộc sống tốt nếu được điều trị phù hợp.
- Nguy cơ kernicterus thấp nhưng vẫn cần theo dõi chặt chẽ.
6.3. Yếu tố ảnh hưởng đến tiên lượng
- Thời điểm chẩn đoán và bắt đầu điều trị.
- Mức độ tuân thủ điều trị của bệnh nhân và gia đình.
- Khả năng tiếp cận với các phương pháp điều trị hiện đại (ví dụ: ghép gan).
- Sự xuất hiện của các biến chứng, đặc biệt là tổn thương thần kinh.
Tài liệu tham khảo
- Erlinger S, et al. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014;146(7):1625-38.
- Bartlett MG, Gourley GR. Assessment of UGT polymorphisms and neonatal jaundice. Semin Perinatol. 2011;35(3):127-33.
- American Academy of Pediatrics. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2022;150(3):e2022058859.
- National Institute for Health and Care Excellence. Jaundice in newborn babies under 28 days. 2023.
- Bosma PJ. Inherited disorders of bilirubin metabolism. J Hepatol. 2003;38(1):107-17.
- van Dijk R, et al. The Crigler-Najjar syndrome: Current perspectives and the application of clinical genetics. Mol Genet Metab. 2015;116(1-2):1-5.
- Jansen PL. Diagnosis and management of Crigler-Najjar syndrome. Eur J Pediatr. 1999;158 Suppl 2:S89-94.
- Schauer R, et al. Treatment options for severe hyperbilirubinemia in neonates: An updated review. World J Pediatr. 2020;16(6):555-564.
- Fagiuoli S, et al. Monogenic diseases that can be cured by liver transplantation. J Hepatol. 2013;59(3):595-612.
- Stevenson DK, et al. Prediction of hyperbilirubinemia in near-term and term infants. Pediatrics. 2001;108(1):31-9.
BÌNH LUẬN