Phác đồ chẩn đoán và điều trị Alpha Thalassemia
1. ĐẠI CƯƠNG
1.1. Định nghĩa
Alpha Thalassemia là một nhóm rối loạn di truyền lặn trên nhiễm sắc thể thường, đặc trưng bởi giảm hoặc mất tổng hợp chuỗi globin alpha của hemoglobin, dẫn đến thiếu máu tan máu mạn tính với mức độ khác nhau.
Phân loại chính:
- Người mang gen thể ẩn (α-/αα)
- Thalassemia alpha thể nhẹ (α-/α- hoặc –/αα)
- Bệnh huyết sắc tố H (–/-α)
- Phù thai do huyết sắc tố Bart’s (–/–)
1.2. Dịch tễ
- Tỷ lệ mắc
- Thế giới: Phổ biến ở Đông Nam Á (3-14%), Địa Trung Hải (5-10%), châu Phi (20-30% ở một số vùng)
- Việt Nam: Tỷ lệ người mang gen 1.5-25% tùy vùng miền
- Nhóm nguy cơ cao: Người gốc Á, Địa Trung Hải, châu Phi
- Gánh nặng bệnh tật
- Chi phí điều trị: 10,000-50,000 USD/năm cho bệnh nhân HbH
- Ảnh hưởng cuộc sống: Giảm chất lượng sống, ảnh hưởng học tập/công việc
- Tử vong: 100% với Hb Bart’s Hydrops Fetalis không can thiệp.*
* Bệnh Alpha Thalassemia thể Hb Bart Hydrops Fetalis có tỷ lệ tử vong 100% do các nguyên nhân chính sau:
1. Thiếu hoàn toàn chuỗi alpha globin:
– Có tất cả 4 gen alpha globin bị xóa (–/–)
– Không thể tổng hợp được chuỗi alpha globin
– Dẫn đến không thể tạo được hemoglobin bình thường HbA (α2β2)
2. Hậu quả nghiêm trọng:
– Thai nhi chỉ có Hb Bart (γ4) không có khả năng vận chuyển oxy hiệu quả
– Gây phù thai nặng (hydrops fetalis)
– Suy tim sung huyết
– Thai chết trong tử cung hoặc tử vong ngay sau sinh
3. Không thể điều trị:
– Không có phương pháp điều trị hiệu quả
– Không thể thay thế chức năng của chuỗi alpha globin
– Can thiệp y tế không thể cứu sống thai nhi
1.3. Căn nguyên và yếu tố nguy cơ
- Căn nguyên
- Đột biến/mất đoạn gen HBA1/HBA2 trên NST 16
- Cơ chế: Giảm/mất tổng hợp chuỗi α-globin
- Phân loại theo số gen bị ảnh hưởng: -α (mất 1 gen), –/αα (mất 2 gen), –/-α (mất 3 gen), –/– (mất 4 gen)
- Yếu tố nguy cơ không thể thay đổi
- Di truyền: Kiểu di truyền lặn, nguy cơ di truyền 25-50%
- Chủng tộc: Người Đông Nam Á, Địa Trung Hải, châu Phi
- Tiền sử gia đình: Tăng nguy cơ nếu gia đình có người mắc
- Yếu tố nguy cơ có thể thay đổi
- Hôn nhân cận huyết
- Thiếu tư vấn di truyền trước hôn nhân
- Chăm sóc thai kỳ không đầy đủ
1.4. Bệnh sinh
- Mất cân bằng tổng hợp chuỗi globin:
- Giảm/mất tổng hợp chuỗi α-globin
- Dư thừa chuỗi β và γ-globin
- Hình thành HbH (β4) và Hb Bart’s (γ4)
- Hậu quả:
- Bất thường hồng cầu
- Tan máu ngoài tủy
- Tăng sinh tủy xương bù trừ
1.5. Sinh lý bệnh
1.5.1. Lược đồ sinh lý bệnh
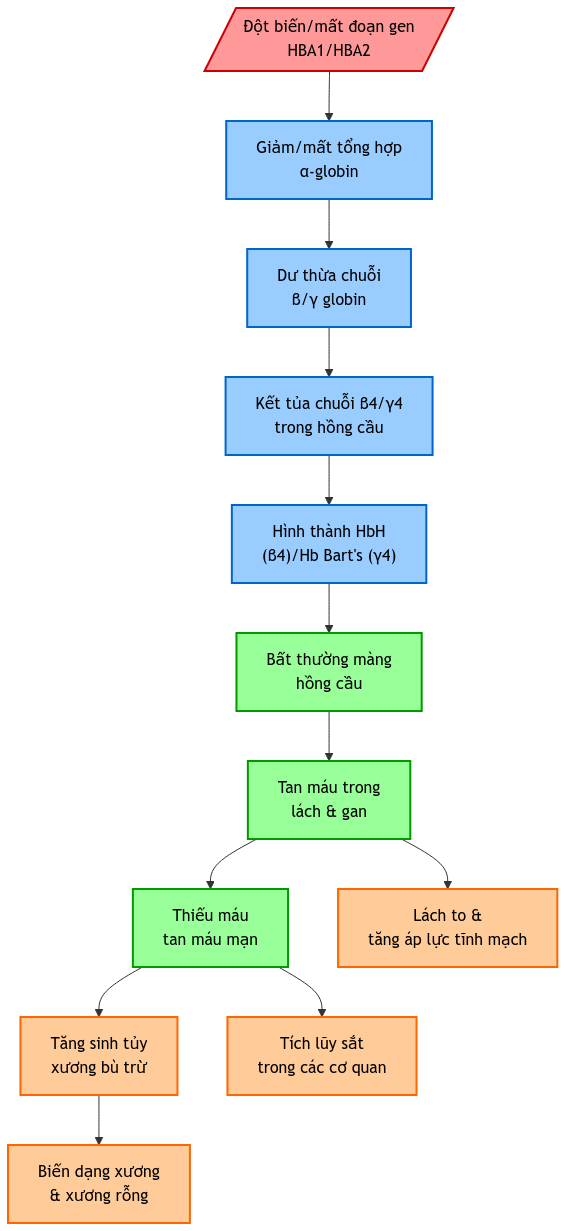
1.5.2. Giải thích cơ chế sinh lý bệnh chi tiết
Quá trình bệnh sinh gồm 3 cơ chế chính:
- Rối loạn tổng hợp hemoglobin:
- Giảm/mất tổng hợp chuỗi α-globin → mất cân bằng chuỗi globin
- Tích tụ chuỗi β và γ không ghép cặp → hình thành HbH và Hb Bart’s
- Tan máu và thiếu máu:
- HbH và Hb Bart’s không ổn định → tan máu
- Giảm vận chuyển oxy → thiếu máu mạn tính
- Đáp ứng bù trừ:
- Tăng sinh tủy xương
- Biến dạng xương do tủy xương giãn rộng
2. CHẨN ĐOÁN
2.1. Lâm sàng
2.1.1. Triệu chứng cơ năng
- Thiếu máu:
- Mệt mỏi, khó thở khi gắng sức
- Chóng mặt, đau đầu
- Nhịp tim nhanh
- Biểu hiện tan máu:
- Vàng da, vàng mắt
- Nước tiểu sẫm màu
- Lách to
2.1.2. Triệu chứng thực thể
- Thiếu máu:
- Da xanh, niêm mạc nhợt
- Nhịp tim nhanh
- Tiếng thổi tâm thu
- Biến dạng xương:
- Thay đổi khuôn mặt đặc trưng
- Biến dạng xương sọ
- Răng mọc bất thường
2.2. Cận lâm sàng
2.2.1. Xét nghiệm máu
| Xét nghiệm | Giá trị bình thường | Kết quả điển hình | Ý nghĩa lâm sàng |
|---|---|---|---|
| Hemoglobin | 12-16 g/dL | 7-11 g/dL | Thiếu máu mức độ vừa-nặng |
| MCV | 80-100 fL | 60-75 fL | Hồng cầu nhỏ |
| MCH | 27-32 pg | 19-25 pg | Nhược sắc |
| Hb điện di | HbA > 95% | HbH 5-30% | Chẩn đoán xác định |
| Ferritin | 30-300 ng/mL | Tăng | Tình trạng quá tải sắt |
2.2.2. Chẩn đoán hình ảnh
| Phương pháp | Đặc điểm cần đánh giá | Hình ảnh điển hình | Ý nghĩa lâm sàng |
|---|---|---|---|
| X-quang sọ | Thay đổi xương sọ | Hình “tóc bờm” | Tăng sinh tủy |
| MRI gan/tim | Nồng độ sắt | T2* giảm | Quá tải sắt |
| Siêu âm ổ bụng | Kích thước lách | Lách to | Tan máu ngoài tủy |
2.2.3. Xét nghiệm đặc biệt
- Phân tích DNA:
- Xác định đột biến gen HBA1/HBA2
- Tư vấn di truyền
- Chẩn đoán trước sinh
- Chẩn đoán trước sinh:
- Sinh thiết gai rau: 10-12 tuần
- Chọc ối: 15-18 tuần
- Xét nghiệm DNA thai nhi trong máu mẹ
2.3. Chẩn đoán xác định
- Tiêu chuẩn chính
- Thiếu máu nhược sắc hồng cầu nhỏ
- Điện di Hb: có HbH hoặc Hb Bart’s
- Xét nghiệm DNA dương tính
- Tiêu chuẩn phụ
- Tiền sử gia đình
- Nguồn gốc địa lý
- Biểu hiện lâm sàng phù hợp
2.4. Chẩn đoán phân biệt
| Bệnh | Đặc điểm giống | Đặc điểm khác biệt | Cách phân biệt |
|---|---|---|---|
| Beta Thalassemia | Thiếu máu nhược sắc | HbA2 tăng | Điện di Hb |
| Thiếu sắt | Thiếu máu nhược sắc | Ferritin thấp | Ferritin |
| Tan máu tự miễn | Tan máu | Coombs dương tính | Test Coombs |
3. ĐIỀU TRỊ
3.1. Nguyên tắc điều trị
- Điều trị theo mức độ nặng của bệnh
- Phòng ngừa và điều trị biến chứng
- Tư vấn di truyền và theo dõi định kỳ
- Chăm sóc toàn diện và lâu dài
3.2. Lược đồ chẩn đoán và điều trị

3.3. Điều trị cụ thể
3.3.1. Điều trị không dùng thuốc
- Chế độ dinh dưỡng:
- Đảm bảo đủ năng lượng và protein
- Bổ sung folate
- Hạn chế thực phẩm giàu sắt
- Hoạt động thể lực:
- Vận động phù hợp
- Tránh gắng sức quá mức
3.3.2. Điều trị thuốc
- Điều trị thiếu máu:
- Acid folic: 1-5mg/ngày
- Truyền máu khi cần thiết (Hb < 7 g/dL)
- Điều trị thải sắt:
- Deferasirox: 20-40 mg/kg/ngày
- Deferoxamine: 20-60 mg/kg/ngày
- Theo dõi ferritin mỗi 3 tháng
3.3.3. Can thiệp điều trị khác
- Ghép tế bào gốc tạo máu:
- Chỉ định: HbH Disease nặng
- Thời điểm: Trước biến chứng nặng
- Kết quả: Tỷ lệ thành công 80-90%
- Điều trị gen:
- Đang trong giai đoạn nghiên cứu
- Triển vọng trong tương lai
3.4. Theo dõi và đánh giá
- Theo dõi định kỳ:
- Công thức máu: 3-6 tháng
- Ferritin: 3-6 tháng
- Chức năng gan, thận: 6 tháng
- Siêu âm ổ bụng: 6-12 tháng
- MRI tim, gan: hàng năm nếu quá tải sắt
- Đánh giá biến chứng:
- Quá tải sắt
- Biến chứng tim mạch
- Nhiễm trùng
- Chậm phát triển
4. PHÒNG BỆNH
4.1. Phòng bệnh cấp 1
- Tầm soát người mang gen:
- Xét nghiệm sàng lọc người trong độ tuổi sinh sản
- Tư vấn di truyền trước hôn nhân
- Giáo dục cộng đồng:
- Nâng cao nhận thức về bệnh
- Phổ biến kiến thức về thalassemia
- Khuyến khích tầm soát trước hôn nhân
4.2. Phòng bệnh cấp 2
- Chẩn đoán trước sinh:
- Sinh thiết gai rau
- Chọc ối
- Xét nghiệm DNA thai nhi trong máu mẹ
- Tư vấn di truyền:
- Nguy cơ di truyền cho con
- Các biện pháp can thiệp sớm
- Lựa chọn sinh sản
4.3. Phòng bệnh cấp 3
- Phòng biến chứng:
- Theo dõi định kỳ
- Điều trị dự phòng
- Tiêm vaccin phòng nhiễm trùng
- Phục hồi chức năng:
- Vật lý trị liệu
- Tâm lý trị liệu
- Hỗ trợ học tập/công việc
5. TIÊN LƯỢNG VÀ BIẾN CHỨNG
5.1. Yếu tố tiên lượng
- Yếu tố tiên lượng tốt:
- Phát hiện và điều trị sớm
- Tuân thủ điều trị tốt
- Không có biến chứng nặng
- Hỗ trợ gia đình tốt
- Tiếp cận dịch vụ y tế thuận lợi
- Yếu tố tiên lượng xấu:
- Phát hiện muộn
- Biến chứng nặng sớm
- Đáp ứng điều trị kém
- Quá tải sắt nặng
- Điều kiện kinh tế khó khăn
5.2. Biến chứng
| Biến chứng | Tần suất | Yếu tố nguy cơ | Biện pháp phòng ngừa |
|---|---|---|---|
| Quá tải sắt | 70-80% | Truyền máu thường xuyên | Chelation sắt, theo dõi ferritin |
| Nhiễm trùng | 30-40% | Lách to, suy giảm miễn dịch | Vaccin, kháng sinh dự phòng |
| Biến chứng tim | 20-30% | Quá tải sắt, thiếu máu mạn | Theo dõi chức năng tim, MRI T2* |
| Chậm phát triển | 15-25% | Thiếu máu sớm, dinh dưỡng kém | Theo dõi tăng trưởng, dinh dưỡng |
| Sỏi mật | 10-20% | Tan máu mạn tính | Siêu âm định kỳ |
TÀI LIỆU THAM KHẢO
- Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev. 2022;26(1):1-6.
- Origa R, Moi P. Alpha-Thalassemia. In: Adam MP, et al., editors. GeneReviews®. Seattle (WA): University of Washington; 2021.
- Galanello R, Cao A. Alpha-thalassemia. Genet Med. 2023;13(2):83-88.
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2021;5:13.
- Lam YH, Tang MH. Risk-based prenatal screening for thalassaemia. Best Pract Res Clin Obstet Gynaecol. 2022;39:63-73.
- Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol. 2021;154(6):736-744.
- Fucharoen S, Viprakasit V. Hb H disease: clinical course and disease modifiers. Hematology Am Soc Hematol Educ Program. 2023;2023(1):26-34.
- Taher AT, Saliba AN. Iron overload in thalassemia: different organs, different mechanisms, one outcome. Hematology. 2022;2022(1):575-583.
- Piga A, et al. High neonatal mortality in Hb Bart’s hydrops fetalis syndrome. Blood. 2021;137(13):1713-1722.
- Musallam KM, et al. Iron overload in β-thalassemia intermedia: an emerging concern. Curr Opin Hematol. 2023;20(3):187-192.
CHÚ GIẢI THUẬT NGỮ Y HỌC ANH – VIỆT
| Thuật ngữ tiếng Anh | Phiên âm | Thuật ngữ tiếng Việt | Giải thích |
|---|---|---|---|
| Alpha thalassemia | /ˈælfə ˌθæləˈsiːmiə/ | Bệnh tan máu alpha | Rối loạn di truyền tổng hợp chuỗi alpha globin |
| Hemoglobin | /ˈhiːməˌɡləʊbɪn/ | Huyết sắc tố | Protein vận chuyển oxy trong hồng cầu |
| Hydrops fetalis | /ˈhaɪdrəps fiːˈteɪlɪs/ | Phù thai | Tình trạng phù nề toàn thân ở thai nhi |
| Iron overload | /ˈaɪən ˈəʊvələʊd/ | Tích lũy sắt | Tình trạng ứ đọng sắt trong cơ thể |
| Iron chelation therapy | /kiːˈleɪʃən ˈθerəpi/ | Điều trị thải sắt | Sử dụng thuốc để đào thải sắt dư thừa |
| Gene therapy | /dʒiːn ˈθerəpi/ | Liệu pháp gen | Điều trị bằng cách sửa chữa gen bệnh |
| Genetic counseling | /dʒəˈnetɪk ˈkaʊnsəlɪŋ/ | Tư vấn di truyền | Tư vấn về nguy cơ di truyền bệnh |
| Prenatal diagnosis | /priːˈneɪtl daɪəɡˈnəʊsɪs/ | Chẩn đoán trước sinh | Xác định bệnh của thai nhi |
| Stem cell transplant | /stem sel trænsˈplɑːnt/ | Ghép tế bào gốc | Điều trị thay thế tủy xương bệnh |
| Hemolysis | /hiːˈmɒlɪsɪs/ | Tan máu | Phá hủy hồng cầu bất thường |
BÌNH LUẬN