Hội chứng mạch vành cấp tính (ACS) đại diện cho phổ lâm sàng của bệnh động mạch vành cấp tính bao gồm đau thắt ngực không ổn định, nhồi máu cơ tim cấp tính (MI) và tử vong đột ngột do bệnh mạch vành. Trong hầu hết các trường hợp, cơ chế cơ bản là tắc nghẽn lưu lượng máu động mạch vành do huyết khối phát triển do sự rò hoặc xói mòn mảng xơ vữa động mạch bên dưới. Các nguyên nhân ít gặp hơn là nhồi máu cơ tim với động mạch vành không tắc nghẽn, bóc tách động mạch vành, co thắt động mạch vành và rối loạn chức năng vi mạch vành.
Chủ đề này tập trung vào các cơ chế của ACS liên quan đến sự mất ổn định của mảng xơ vữa động mạch.
Tác giả: Filippo Crea, MDFrank Kolodgie, PhDAloke Finn, MDRenu Virmani, MD;
Dịch: Ths.Bs. Lê Đình Sáng – Bệnh viện Hữu nghị Đa khoa Nghệ An
SỰ HÌNH THÀNH VÀ TIẾN TRIỂN CỦA MẢNG XƠ VỮA ĐỘNG MẠCH
Mảng xơ vữa động mạch hiện diện ở hầu hết bệnh nhân mắc ACS. Sự hình thành xơ vữa động mạch là một quá trình năng động. Các giai đoạn tiến triển đã được mô tả (hình 1A-B) [1,2].

Hình 1A. Sự tiến triển của chứng xơ vữa động mạch vành ở người I

Hình 1B. Sự tiến triển của chứng xơ vữa động mạch vành ở người II
Xanthoma nội mạc thích ứng (vệt mỡ) và dày lên nội mạc — Xanthoma nội mạc thích ứng (vệt mỡ) và dày nội mạc được coi là biểu hiện sớm nhất của bệnh xơ vữa động mạch.
“Xanthoma” là một thuật ngữ bệnh lý chungmô tả sự tích tụ tập trung của các đại thực bào chứa đầy lipid bên trong nội mạc. Ở người, xanthomata được biết là sẽ thoái triển, vì sự phân bố của các tổn thương ở thập kỷ thứ ba của cuộc đời trở về sau rất khác so với những vệt mỡ này được nhìn thấy ở trẻ em [3,4]. Các vệt mỡ sớm (xanthoma nội mạc), chủ yếu được quan sát thấy ở các điểm nhánh, tương ứng với sự tích tụ của các đại thực bào trong nội mạc.
Dày nội mạc, một quá trình không gây xơ vữa liên quan đến tế bào cơ trơn, xảy ra ở trẻ em ở những vị trí tương tự như mảng xơ vữa tiến triển ở người lớn. Về mặt mô học, sự dày lên của nội mạc bao gồm chủ yếu là các tế bào cơ trơn trong ma trận proteoglycan-collagen với rất ít hoặc không có tế bào viêm xâm nhập. 30% trẻ sơ sinh có biểu hiện dày lên ở nội mạc, đặc biệt là ở các điểm phân nhánh nhưng đến tháng thứ sáu, hầu hết tất cả đều có biểu hiện dày lên ở nội mạc. Sự hiểu biết của chúng ta về cơ chế sinh lý bệnh của sự phát triển của chúng vẫn còn rất hạn chế.
Giai đoạn vượt quá xanthoma nội mạc/dày nội mạc là mảng bám tiến triển hơn được gọi là “dày nội mạc bệnh lý” và được đặc trưng bởi các bể lipid ngoại bào chứa proteoglycan mà không bị hoại tử [1,5]. Các nhóm lipid bao gồm các khu vực giàu hyaluronan và proteoglycan nhưng không có tế bào cơ trơn và tế bào viêm; tuy nhiên, các nhóm lipid rất giàu lipid ngoại bào. Các bể lipid có xu hướng phát triển ở các lớp nội mạc sâu hơn gần lớp áo giữa của động mạch. Khi có mặt, đại thực bào và thâm nhiễm tế bào lympho T ở gần bề mặt lòng ống và bị hạn chế khỏi khu vực tích lũy lipid.
Tình trạng vôi hóa sớm thường được quan sát thấy ở các khu vực chứa lipid và có thể là kết quả của sự chết của các tế bào cơ trơn [6].
Viêm và tiến triển mảng bám – Xơ vữa động mạch đã được xác định là một bệnh viêm mãn tính, với sự thâm nhiễm bạch cầu đơn nhân là một trong những bước đầu [7]. Sự hiện diện của lipoprotein nội mạc và các sản phẩm có nguồn gốc hoặc biến đổi của chúng gây ra sự gia tăng biểu hiện của các phân tử bám dính trên bề mặt nội mô. Sự kết dính của các tế bào viêm liên quan đến sự biểu hiện của các selectin, tạo điều kiện thuận lợi cho sự gắn kết chắc chắn của bạch cầu đơn nhân với các integrin nội mô và di chuyển bằng các protein nối nội mô [8].
Quá trình oxy hóa các hạt lipoprotein mật độ thấp (LDL) có thể tham gia vào quá trình khởi đầu và tiến triển của chứng xơ vữa động mạch và được thúc đẩy bởi các đại thực bào, tế bào nội mô và tế bào cơ trơn [9,10]. Quá trình oxy hóa LDL có thể xảy ra do lipoxygenase, axit hypochlorous được tạo ra bởi myeloperoxidase, oxit nitric được tạo ra bởi synthase oxit cảm ứng, anion superoxide được tạo ra bởi NADPH oxyase và từ sắt có nguồn gốc từ heme [11,12]. Các loại oxy phản ứng như vậy có nguồn gốc từ đại thực bào, tế bào nội mô và tế bào cơ trơn. LDL bị oxy hóa thúc đẩy sự hấp dẫn hóa học, ví dụ, bằng cách tạo ra sự tiết ra protein-1 đại thực bào-hóa học (MCP-1) bởi các tế bào nội mô [12,13]. Hơn nữa, đại thực bào biểu hiện một số thụ thể nhặt rác, có thể liên kết nhiều phối tử, bao gồm lipoprotein biến đổi, lipoprotein tự nhiên và phospholipid anion, nhiều trong số đó tạo điều kiện cho sự tích tụ lớn của este cholesterol nội bào và cholesterol tự do [11].
Các đại thực bào và tế bào nội mô cũng biểu hiện các thụ thể giống Toll (TLR) trên bề mặt tế bào của chúng và chứa các phân tử bộ điều hợp khác nhau (đặc biệt là MyD88) tham gia truyền tín hiệu xuôi dòng [14]. TLR kích hoạt yếu tố hạt nhân của yếu tố phiên mã tiền viêm kappa-B, dẫn đến việc sản xuất các cytokine làm tăng tình trạng viêm cục bộ và tăng sinh tế bào cơ trơn [15,16]. Các nghiên cứu in vitro đã chỉ ra rằng sự biểu hiện cơ bản của TLR-4 bởi các đại thực bào được điều chỉnh tăng lên bởi LDL bị oxy hóa [17] và sự hấp thu lipid bị oxy hóa vào đại thực bào được tạo điều kiện thuận lợi bởi một dị vòng CD36/TLR4/TLR6 khởi tạo tín hiệu TLR4 [18]. Những dữ liệu này cung cấp mối liên hệ sinh lý bệnh tiềm ẩn giữa sự tích tụ lipid, các cytokine gây viêm và xơ vữa động mạch.
Fibroatheroma – Fibroatheroma, còn được gọi là mảng xơ vữa nắp xơ, được coi là đại diện cho dạng tổn thương xơ vữa động mạch tiến triển đầu tiên [1,19]. Tổn thương này được đặc trưng bởi sự hiện diện của lõi hoại tử được tạo ra do sự xâm nhập của đại thực bào vào các bể lipid và được bao bọc bởi mô sợi xung quanh (nắp sợi). Phần lõi hoại tử là do cái chết của các đại thực bào chứa đầy lipid, còn được gọi là tế bào bọt và lipid có nguồn gốc từ huyết tương [20,21]. Tế bào bọt chứa este cholesterol và cholesterol tự do. Khi mảng bám phát triển, hàm lượng cholesterol tự do của tổn thương mảng bám tăng lên, cũng như tỷ lệ cholesterol tự do và ester hóa [22,23]. Tỷ lệ cholesterol tự do và phospholipid cao trong màng tế bào đã được chứng minh là gây độc cho tế bào, và do đó, độc tế bào do cholesterol tự do gây ra có thể góp phần gây hoại tử tế bào bọt, cần phân biệt với apoptosis, là một tế bào được lập trình tự nhiên. tử vong dẫn đến lõi hoại tử giãn nở (hình 2) [24].
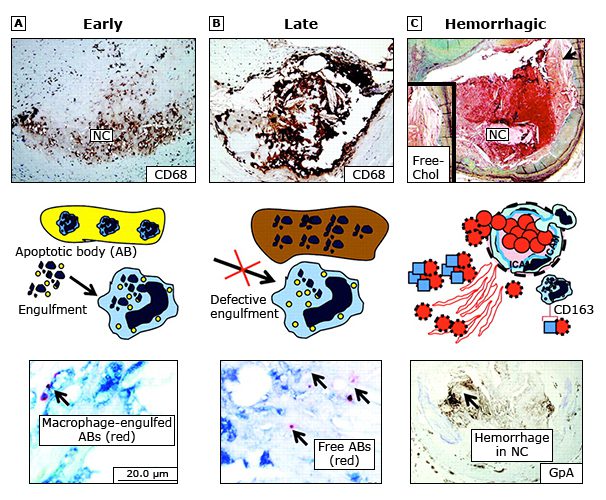
(Các) cơ chế giả định về sự mở rộng lõi hoại tử trong các mảng vành của con người. Từ A đến C lần lượt minh họa khái niệm hoại tử sớm, hoại tử muộn và hoại tử xuất huyết. Một nhóm lipid giàu proteoglycan và đại thực bào dương tính với CD68 là đặc điểm của u xơ xơ tử cung sớm; nói chung là thiếu các tế bào cơ trơn.
(A) Các đại thực bào xâm nhập có khả năng nhấn chìm các thể apoptotic (AB) được nhận biết rõ ràng trong lõi hoại tử (NC).
(B) Khi tổn thương tiến triển, hoại tử muộn được biểu hiện bằng sự gia tăng số lượng đại thực bào chết và sự ly giải tế bào cũng như sự mất mát rõ ràng của chất nền ngoại bào. Trong trường hợp này, người ta thường thấy các thể apoptotic tự do, có thể cho thấy khả năng đào thải (sự sủi bọt) bị khiếm khuyết của các đại thực bào thường trú.
(C) Trong ví dụ thứ ba, xuất huyết có thể thúc đẩy sự mở rộng tương đối nhanh chóng của lõi hoại tử, nơi màng hồng cầu cung cấp cholesterol tự do (Free-Chol, mũi tên), có thể gây viêm thứ phát. Các đại thực bào thường trú có khả năng loại bỏ các phức hợp hemoglobin/haptoglobin thông qua thụ thể CD163, mặc dù hiệu quả của cơ chế này cũng có thể bị ảnh hưởng.
Hình 2. Cơ chế giãn nở lõi hoại tử.
(From: Kolodgie F. Pathogenesis of atherosclerosis and the unstable plaque. In: Kwiterovich J. The Johns Hopkins Textbook of Dyslipidemia, Lippincott Williams & Wilkins, Philadelphia 2009. Copyright © 2009 Lippincott Williams & Wilkins)
Trong khi sự tích tụ của các đại thực bào bọt chứa đầy lipid có tác động quan trọng đến sự phát triển của tổn thương xơ vữa động mạch, thì các đại thực bào này có thể chết chủ yếu thông qua các cơ chế liên quan đến quá trình tự hủy.
Mặc dù cái chết của đại thực bào xảy ra trong tất cả các giai đoạn của chứng xơ vữa động mạch, đại thực bào theo chương trình được quan sát thường xuyên hơn ở các tổn thương xơ vữa động mạch tiến triển [25]. Các nghiên cứu in vivo mở rộng cho thấy rằng hiệu quả thanh thải thực bào của các tế bào chết theo chương trình (thường được gọi là loại bỏ tế bào theo chương trình hoặc tăng bọt) có thể hỗ trợ trong các tổn thương sớm với ảnh hưởng tối thiểu đến tế bào tổn thương, trong khi đối với các tổn thương muộn, đại thực bào có thể biểu hiện khả năng thanh thải thực bào khiếm khuyết , trong đó tàn dư của các tế bào chết có thể dẫn đến kích hoạt các phản ứng tiền viêm và tiếp theo là sự mở rộng lõi hoại tử và tiến triển tổn thương (hình 2) [26,27]. Có khả năng là cả độc tế bào tế bào gây ra bởi cholesterol tự do (đã thảo luận ở trên) và việc loại bỏ khiếm khuyết các tế bào chết theo chương trình đều góp phần làm cho lõi hoại tử mở rộng.
Xuất huyết trong mảng bám là một yếu tố quan trọng khác góp phần vào việc mở rộng lõi hoại tử. Mặc dù các đại thực bào chết theo chương trình có thể là nguồn cung cấp cholesterol tự do trong quá trình phát triển và mở rộng của lõi hoại tử, nhưng không chắc rằng tổng lượng cholesterol tự do trong các mảng bám chỉ xuất phát từ các tế bào bọt, vì một phần lớn cholesterol trong các tế bào bọt được este hóa. Một số tinh thể cholesterol trong tổn thương có nguồn gốc từ hồng cầu, vì chúng thường được quan sát thấy ở các vị trí xuất huyết [28]. Ngoài ra, hàm lượng cholesterol trong màng hồng cầu là loại cholesterol tự do giàu nhất trong tất cả các tế bào trong cơ thể. Xuất huyết trong lõi hoại tử là một hiện tượng phổ biến, do đó có khả năng cholesterol tự do trong lõi hoại tử cũng đến từ màng tế bào hồng cầu (hình 2 và hình 1).
Nguồn gốc của xuất huyết có lẽ là do rò rỉ mạch máu, xâm nhập vào mảng bám chủ yếu từ lớp vỏ ngoài để đáp ứng với môi trường thiếu oxy được tạo ra bởi sự gia tăng gánh nặng tổn thương và các đại thực bào viêm. Động mạch nuôi mạch máu (vasa vasorum) trong các mảng thường thiếu màng đáy nguyên vẹn, kém ổn định bởi các tế bào ngoại vi xung quanh và biểu hiện các mối nối nội mô kém, có khả năng là nguyên nhân dẫn đến sự thiếu tính toàn vẹn của chúng [29].
Ở những vùng xuất huyết mảng bám, sự ly giải hồng cầu dẫn đến giải phóng huyết sắc tố tự do. Sự hấp thu huyết sắc tố: phức hợp haptoglobin thông qua thụ thể CD163 biểu hiện trên các đại thực bào cho phép biệt hóa các tế bào này thành kiểu hình tế bào không bọt duy nhất được đặc trưng bởi sự thiếu khả năng giữ lipid và giảm biểu hiện của các cytokine gây viêm như TNF alpha. Những tế bào này đóng một vai trò quan trọng trong việc giải độc sắt có nguồn gốc từ huyết sắc tố và do đó được cho là có tác dụng bảo vệ xơ vữa [30,31]. Tuy nhiên, bằng chứng chỉ ra rằng những tế bào này đóng vai trò gây bệnh trong chứng xơ vữa động mạch bằng cách thúc đẩy sự hình thành mảng bám, tính thấm của mạch máu và tuyển dụng tế bào viêm thông qua việc giải phóng yếu tố tăng trưởng nội mô mạch máu A [32].
Trong các u xơ xơ muộn, người ta quan sát thấy các tập hợp mảnh vụn tế bào rời rạc, tăng cholesterol tự do và gần như cạn kiệt hoàn toàn chất nền ngoại bào. Mảng xơ vữa nắp xơ có thể phát triển thành tổn thương với mức độ hẹp lòng mạch đáng kể sau các đợt xuất huyết có hoặc không có vôi hóa và tổn thương bề mặt. Trong khu vực lõi hoại tử, sự vôi hóa được quan sát dưới dạng các vùng có dấu lấm chấm (>15 micromet) do tế bào đại thực bào chết. Các vi vôi hóa như vậy kết hợp lại để tạo thành các khối lớn hơn có thể liên quan đến cả lõi hoại tử và ma trận ngoại bào giàu collagen xung quanh để tạo thành các đốm và mảnh vôi hóa [33].
Tái tạo động mạch vành – Sự gia tăng kích thước tổn thương làm ảnh hưởng đến lưu lượng máu ở lòng mạch chỉ khi quan sát thấy mặt cắt ngang của lòng mạch thu hẹp từ 40% trở lên. Sự vắng mặt của tình trạng mất lòng mạch không phụ thuộc vào gánh nặng tổn thương ở các mảng ban đầu có liên quan đến sự giãn nở bù trừ của mạch máu, tức là tái cấu trúc động mạch vành dương tính, thường được gọi là “hiện tượng Glagov” [34,35].
Các nghiên cứu siêu âm nội mạch và khám nghiệm tử thi đã tiết lộ rằng việc tái cấu trúc tích cực của thành động mạch vành có liên quan đến hàm lượng lipid cao hơn và thâm nhiễm đại thực bào, cũng như các đặc điểm mảng bám không ổn định như vỡ cấp tính, xuất huyết trong mảng bám và Mảng xơ vữa nắp mỏng [36,37]. Ngược lại, các tổn thương xói mòn hoặc tắc nghẽn toàn bộ mãn tính biểu hiện sự tái cấu trúc tiêu cực [36].
Mảng xơ vữa nắp mỏng – Mảng xơ vữa nắp mỏng được định nghĩa là một tổn thương báo trước sự vỡ mảng bám, có lõi hoại tử và nắp mỏng và bị vỡ với huyết khối ở lòng quá mức. Các mũ bị vỡ được đo trong các trường hợp khám nghiệm tử thi và có độ dày 23±19 micron tại vị trí vỡ, và do đó, từ các phép đo này, một khối u xơ vữa dạng mũ mỏng được xác định không chỉ có lõi hoại tử mà còn phải <65 micron vì khoảng tin cậy 95% được xác định là 64 micron. Do đó, để mô tả rõ hơn u xơ xơ vỏ mỏng, tổn thương tiền thân của vỡ nắp, độ dày phải <65 micron.
U xơ vữa động mạch dường như có thể là tổn thương tiền thân của mảng xơ vữa nắp mỏng, mặc dù dữ liệu in vivo rõ ràng cho thấy điều này chưa được chứng minh. Có thể các mảng xơ vữa nắp mỏng được hình thành mới.
Có thể ở một số bệnh nhân, nắp sợi trở nên mỏng do hoạt động chiếm ưu thế của ma trận metallicoproteinase, được giải phóng từ các đại thực bào được kích hoạt, nằm bên dưới nắp sợi giàu collagen. Ngược lại, ở những bệnh nhân khác, độ dày của vỏ xơ có thể tăng lên do sự tổng hợp collagen chiếm ưu thế. Vì vậy, sự tiến triển của tổn thương động mạch vành khá năng động [38]. Mảng xơ vữa nắp mỏng, còn được gọi là “mảng bám dễ bị tổn thương”, được xác định bởi một lõi hoại tử lớn (chiếm khoảng 25% diện tích mảng bám) được ngăn cách với lòng bằng một nắp sợi mỏng, độ dày dưới 65 micromet. Mũ xơ bị thâm nhiễm nhiều bởi các đại thực bào và ở mức độ thấp hơn là tế bào lympho T (hình 2) [39]. Thông thường, phần mũ xơ có rất ít hoặc không có tế bào cơ trơn. Bởi vì nó giống với các mảng đã vỡ về hình thái (trừ phần gián đoạn của nắp), tổn thương có đặc điểm rõ ràng này được coi là khúc dạo đầu cho sự vỡ mảng bám [1,40]. Phải thừa nhận rằng dữ liệu về nguyên nhân và kết quả bị thiếu trong mô hình này bởi vì chúng tôi không thể phát hiện mảng bám dễ bị tổn thương ở người in vivo mà cuối cùng bị vỡ ra một cách nhất quán và vì phần lớn các mô hình động vật bị vỡ mảng bám đều bị thiếu.
Mặc dù có những điểm tương đồng về hình thái với các tổn thương có mảng bám bị vỡ, các Mảng xơ vữa nắp mỏng biểu hiện xu hướng lõi hoại tử nhỏ hơn, ít đại thực bào nắp hơn và ít vôi hóa hơn so với khi vỡ [41]. Thu hẹp lòng mạch cắt ngang ở u xơ xơ vữa nắp mỏng thường ít hơn so với hẹp lòng cơ bản trong vỡ cấp tính [41]. Phần lớn các Mảng xơ vữa nắp mỏng (trên 80%) ở nạn nhân đột tử do mạch vành có <75% mặt cắt ngang thu hẹp lòng ống (hoặc hẹp đường kính <50%) [42].
Độ dày nắp sợi <65 micromet là định nghĩa được chấp nhận rộng rãi về nắp sợi mỏng. Tuy nhiên, do việc cố định và xử lý mô dẫn đến sự co rút khoảng 25 đến 40 phần trăm diện tích thành động mạch ở động mạch vành ở người bị xơ vữa động mạch từ trung bình đến nặng [43], nên nó có thể không phải là giá trị ngưỡng thích hợp để đánh giá tình trạng vỡ- dễ bị mảng bám in vivo. Trên thực tế, các nghiên cứu chụp cắt lớp mạch lạc quang học đánh giá bệnh nhân bị vỡ mảng bám đã chứng minh rằng độ dày vỏ xơ bị phá vỡ <70 micromet chỉ được quan sát thấy ở 67% [44] và giá trị trung bình (phạm vi tứ phân vị) của độ dày vỏ là 54 (50 đến 60) micromet, với 95% độ dày nắp mỏng nhất nhỏ hơn 80 micromet [45].
VỠ MẢNG BÁM
Hầu hết ACS là do mất tính toàn vẹn của lớp vỏ bảo vệ trên mảng xơ vữa động mạch. Điều này xảy ra khi mảng bám bị vỡ khi lớp xơ phủ trên mảng bám bị phá vỡ hoặc bị xói mòn khi lớp nội mô của mảng bám bị xáo trộn. Sự phá vỡ lớp vỏ bảo vệ này cho phép máu tiếp xúc với các chất gây huyết khối cao trong lõi hoại tử (bao gồm cả yếu tố mô) [1,46].
Tại vị trí vỡ, huyết khối trong lòng thường giàu tiểu cầu, do đó tạo ra màu trắng đục (huyết khối màu trắng), trong khi ở đầu gần và xa gần vị trí lan truyền của huyết khối, nó xuất hiện màu đỏ (huyết khối màu đỏ). , vì nó bao gồm các lớp fibrin và hồng cầu. Theo thời gian, quá trình lành vết thương do huyết khối được đặc trưng bởi sự xâm nhập của các tế bào cơ trơn, tích lũy các protein ma trận ngoại bào (tức là proteoglycan và collagen), tân mạch, viêm và tái tạo bề mặt lòng mạch. Sự phá vỡ nắp xơ cho phép các thành phần tế bào và phi tế bào của máu tiếp xúc trực tiếp với các thành phần gây huyết khối cao của lõi hoại tử và được cho là nguyên nhân trực tiếp cho sự phát triển thực sự của huyết khối. Về mặt lịch sử, lõi hoại tử được cho là nguồn cung cấp yếu tố mô chính; tuy nhiên, hiện nay người ta tin rằng các bạch cầu đơn nhân tuần hoàn, thay vì chỉ có đại thực bào, cung cấp các yếu tố mô kích hoạt và lan truyền huyết khối cấp tính nằm trên chứng xơ vữa động mạch vành không ổn định [47].
Mặc dù cơ chế chính xác của việc vỡ mảng bám chưa được hiểu rõ, nhưng người ta chấp nhận rộng rãi rằng sự phá vỡ xảy ra ở vị trí của mũ xơ bị các đại thực bào và tế bào lympho T xâm nhập mạnh, nơi lõi hoại tử bên dưới thường lớn [1,7] và có liên quan đến việc kích hoạt hệ thống miễn dịch thích ứng [48]. Trong khi sự mở rộng lõi hoại tử và tái cấu trúc tích cực có thể liên quan đến sự tiến triển của mảng bám và tổn thương tổn thương, sự thoái hóa thực sự của nắp xơ được cho là xảy ra thông qua sự phân hủy các protein ma trận ngoại bào bởi các ma trận metallicoproteinase (MMP) được tiết ra [49]. Điều này, cùng với các yếu tố quan trọng khác như apoptosis (chết tế bào theo chương trình) và lực lưu biến cục bộ bao gồm co thắt mạch, có liên quan đến sự phát triển của vỡ mảng bám.
Các tế bào cơ trơn mạch máu tổng hợp các protein ma trận ngoại bào cần thiết như collagen và Elastin từ các axit amin. Collagen dạng sợi, đặc biệt là collagen loại I, cung cấp phần lớn độ bền kéo cho nắp sợi. Quá trình tổng hợp collagen có thể bị ức chế bởi gamma interferon do tế bào T kích hoạt tiết ra (hình 3) [50]. Hơn nữa, việc kích hoạt tế bào T dẫn đến sự biểu hiện của các phối tử CD40 (CD40L/CD154), liên kết với các thụ thể CD40 trên đại thực bào, tế bào lympho B và các tế bào khác bao gồm tế bào nội mô và tế bào cơ trơn [7,51-53]. Sự biểu hiện của CD40L trên tế bào T thúc đẩy quá trình phân giải protein mô thông qua việc giải phóng MMP.
Sự mất mát collagen dạng sợi do các MMP hoạt hóa được cho là nguyên nhân làm mỏng mũ xơ và do đó có thể đóng vai trò then chốt trong sự phát triển của vỡ mảng bám (hình 3). Khe phân giải protein ban đầu trong chuỗi collagen được cung cấp bởi collagenase MMPs-1, -8 và -13, trong khi gelatinase MMP-2 và -9 hỗ trợ sự phân hủy thêm các mảnh collagen [7,54-57]. Mặt khác, động mạch cũng có chất đối kháng nội sinh với MMP, chất ức chế mô của metallicoproteinase [8]. Các mảng xơ vữa biểu hiện sự phân cắt collagen loại I tại các vị trí có nhiều đại thực bào biểu hiện cả MMP-1 và -13 [57]. Các proteinase khác có khả năng phân hủy chất nền ngoại bào bao gồm họ cathepsin (cathepsin S và K) và chất ức chế Cysatin C [58]. Không giống như sự phân hủy collagen bởi MMP, hoạt động phân hủy chất đàn hồi có liên quan nhiều hơn đến việc tái cấu trúc và di chuyển và tăng sinh tế bào [55]. Trong khi sự phân hủy độ đàn hồi có thể quan trọng hơn trong việc hình thành chứng phình động mạch, thì sự phân hủy collagen có thể là yếu tố chính quyết định sự vỡ mảng bám [59].
Chết tế bào theo chương trình cũng có thể đóng một vai trò quan trọng trong sự phát triển của vỡ mảng bám [60]. Đại thực bào và tế bào cơ trơn bị chết theo chương trình đã được quan sát thấy trong cả quá trình tiến triển và hồi phục của mảng xơ vữa động mạch [61]. Các vị trí vỡ mảng bám thường có ít hơn hoặc không có tế bào cơ trơn, là những tế bào chính cần thiết cho quá trình tổng hợp protein ma trận ngoại bào và duy trì nắp sợi. Các nghiên cứu in vitro đã chỉ ra rằng các chất trung gian khác nhau được tiết ra bởi đại thực bào và tế bào lympho T, bao gồm interferon-gamma, phối tử Fas, yếu tố hoại tử khối u-a, interleukin-1 và các loại oxy phản ứng, có thể thúc đẩy quá trình apoptosis của tế bào cơ trơn [62], mà có thể giải thích cho việc giảm số lượng tế bào cơ trơn gặp trong xơ vữa động mạch nắp mỏng và các mảng vỡ [63].
Mặc dù chết tế bào theo chương trình của đại thực bào thường được quan sát thấy ở các vị trí vỡ mảng bám, nhưng vẫn chưa biết liệu chết tế bào theo chương trình đơn thuần có khả năng gây ra biến cố chính hay không [64]. Các nghiên cứu in vitro đã xác định được các chất trung gian mạnh, chẳng hạn như lipoprotein mật độ thấp bị oxy hóa, có khả năng gây ra chết tế bào theo chương trình của đại thực bào [60]. Gamma INF đã được chứng minh là gây ra cái chết tế bào theo chương trình của đại thực bào THP-1 [61], đồng thời nó cũng dẫn đến sự tổng hợp thêm protein-1 chất hóa học thu hút đại thực bào, có thể thúc đẩy các phản ứng viêm bổ sung.
Về mặt hình thái, một mảng bám bị vỡ cho thấy có huyết khối trong lòng ống nằm trên một lớp xơ mỏng bị phá vỡ, bị các đại thực bào và tế bào lympho T xâm nhập [65]. Huyết khối tiếp xúc với lõi hoại tử bên dưới (hình 3). Mũ sợi bao gồm chủ yếu là collagen loại I với các đại thực bào và tế bào lympho ở các mức độ khác nhau, trong khi thành phần tế bào cơ trơn bên trong mũ không có hoặc thưa thớt. Ở những tổn thương đã vỡ, độ dày trung bình của vỏ xơ là 23±19 micromet, với 95% vỏ có kích thước nhỏ hơn 65 micromet [39]. Mặc dù người ta đã chấp nhận rộng rãi rằng vỡ mũ xơ xảy ra ở điểm yếu nhất của nó, thường là gần vùng vai, các nghiên cứu khám nghiệm tử thi sử dụng các phần cắt nối tiếp chứng minh số lượng đứt bằng nhau xảy ra ở phần giữa của mũ xơ [46]. Ở một số bệnh nhân, vỡ mảng bám có thể liên quan đến sự kết tinh cholesterol đột ngột bên trong mảng bám dễ bị vỡ, nhưng cần nghiên cứu thêm để xác nhận rằng điều này thực sự xảy ra trong cơ thể [66].
Vết nứt mảng bám được quan sát thấy ở <10% tổn thương và thường liên quan đến hoạt hóa viêm toàn mạch vành và bằng chứng toàn thân về hoạt hóa miễn dịch bẩm sinh và thích nghi [67,68]. Đáng chú ý, các nghiên cứu khám nghiệm tử thi các nạn nhân tử vong đột ngột do mạch vành đã chỉ ra rằng khoảng 40% các mảng vỡ xảy ra ở các vị trí tổn thương có đường kính hẹp dưới 50% [69].
Các nghiên cứu về cơ sinh học và hình ảnh đã xem xét vai trò của ứng suất biến dạng huyết động trong việc làm mất ổn định các mảng bám dễ bị tổn thương [70-72]. Các khu vực có áp suất “vòng” theo chu vi cao và dòng chảy bị xáo trộn có thể góp phần làm vỡ mảng bám. Ứng suất cắt cao hơn (bình thường so với ứng suất theo chu vi, song song với bề mặt nội mạc) bảo vệ thành động mạch khỏi sự hình thành tổn thương và biến chứng. Ứng suất cắt ảnh hưởng đến các quá trình chi phối hình thái và thành phần của mũ sợi [73], trong đó ứng suất chu vi cực đại tăng lên lớn hơn ở các mũ sợi mỏng hơn [70]. Các vùng có ứng suất chu vi cao thường biểu hiện biến dạng cao [72] và khi áp dụng vào phần mũ xơ bị suy yếu, có thể dẫn đến vỡ, đặc biệt khi có sự vôi hóa vi mô [74,75].
Các mảng bám dễ bị tổn thương và tình trạng vỡ trong tương lai — Những nỗ lực sâu rộng đã được dành để xác định những “mảng bám dễ bị tổn thương” dễ bị vỡ này [1,76-78]. Những tiến bộ trong công nghệ hình ảnh cho phép chúng tôi đánh giá một số đặc điểm hình thái của mảng xơ vữa liên quan đến sự phát triển của ACS in vivo, trong khi các chiến lược phát hiện và điều trị các mảng xơ vữa dễ bị tổn thương để ngăn ngừa vỡ mảng xơ vữa cần được cải tiến thêm.
Chụp cắt lớp vi tính (CT) động mạch đang nổi lên như một phương pháp đầy hứa hẹn để đánh giá không xâm lấn tình trạng hẹp động mạch vành và đặc điểm mảng bám [79-83].
Một nghiên cứu lâm sàng đã báo cáo tính hữu ích tiềm tàng của chụp CT động mạch để xác định các mảng bám dễ bị tổn thương chưa vỡ ở những bệnh nhân chưa trải qua biến cố mạch vành cấp tính [84]. Trong tổng số 1059 bệnh nhân được chụp CT mạch, hai đặc điểm quan trọng của CT, tái cấu trúc dương tính và mảng bám đậm độ thấp, được xác định ở 45 bệnh nhân, và ở 27 bệnh nhân khác chỉ có một đặc điểm. Trong số 45 bệnh nhân có hai đặc điểm này, 10 bệnh nhân (22,2%) đã phát triển ACS trong thời gian theo dõi trung bình là 27 tháng, trong khi một bệnh nhân (3,7%) có một đặc điểm duy nhất đã phát triển ACS. Ngược lại, trong số 820 bệnh nhân âm tính với cả hai đặc điểm, chỉ có 4 (0,5%) phát triển ACS. Nghiên cứu này cho thấy các tổn thương mạch vành cho thấy cả sự tái cấu trúc dương tính và các mảng đậm độ thấp trên chụp CT mạch đều có nguy cơ cao hơn mắc các biến cố mạch vành cấp tính trong tương lai. Tuy nhiên, giá trị tiên đoán dương tính rất thấp của mảng bám dễ bị tổn thương đối với ACS tiếp theo trong nghiên cứu này và trong các nghiên cứu tương tự khác [85] khiến phương pháp này có giá trị lâm sàng hạn chế cũng do tiếp xúc với bức xạ, sử dụng chất cản quang và chi phí.
Những lý do dẫn đến hiệu quả dự đoán thấp của việc xác định mảng bám dễ bị tổn thương đã được làm rõ bằng nghiên cứu Cung cấp các quan sát khu vực để nghiên cứu các yếu tố dự đoán sự kiện ở cây mạch vành (PROSPECT), một nghiên cứu tiền cứu đa trung tâm, trong đó 697 bệnh nhân mắc ACS đã được ghi danh. Những bệnh nhân này đã được chụp động mạch vành ba mạch và chụp ảnh siêu âm nội mạch thang màu xám và tần số vô tuyến sau khi can thiệp mạch vành qua da [86]. Tỷ lệ tích lũy trong ba năm của các biến cố tim mạch nặng (tử vong do nguyên nhân tim, ngừng tim, nhồi máu cơ tim hoặc tái nhập viện do đau thắt ngực không ổn định hoặc tiến triển) cũng có thể quy cho sự tái phát tại vị trí tổn thương thủ phạm (13%). đối với các tổn thương không phải do thủ phạm (12%). Hơn nữa, trong số 595 u xơ vữa vỏ mỏng ở các tổn thương không phải thủ phạm lúc ban đầu, các biến cố tim mạch bất lợi chính chỉ xảy ra ở 26 tổn thương (5%) với thời gian theo dõi trung bình là 3,4 năm (tỷ lệ biến cố Kaplan-Meier ước tính là 4,9%). Điều này có lẽ là do phần lớn các mảng bám dễ bị tổn thương sẽ lành lại [38] và do các kỹ thuật hình ảnh không thể xác định được các mảng bám sẽ gây ra ACS do xói mòn hoặc thay đổi chức năng mạch vành. Các công nghệ khác, chẳng hạn như quang phổ cận hồng ngoại, cũng đã được sử dụng để xác định các mảng lõi lipid, như đã báo cáo trong thử nghiệm đa trung tâm PROSPECT II (Cung cấp các quan sát khu vực để nghiên cứu các yếu tố dự đoán sự kiện trong cây mạch vành II) [87].
Chữa lành mảng bám – Các nghiên cứu lâm sàng sử dụng hình ảnh nội mạch đã gợi ý rằng phần lớn các u xơ vữa dạng mỏng có xu hướng lành lại theo thời gian, trong khi một phần nhỏ hơn là nguyên nhân gây ra ACS khi theo dõi [38].
Các tổn thương đã lành xảy ra tại các vị trí bị vỡ trước đó với sự hình thành huyết khối, có thể có hoặc không liên quan đến các triệu chứng. Chúng được định nghĩa là loại mảng xơ vữa động mạch thứ ba, hai loại còn lại là mảng không tiến triển và dễ tiến triển.
Các tổn thương đã lành được xác định dựa trên cơ sở vỏ xơ bị phá vỡ với phản ứng sửa chữa bao gồm các tế bào cơ trơn được bao quanh bởi proteoglycan và/hoặc chất nền giàu collagen, có hoặc không có fibrin tùy thuộc vào giai đoạn lành thương [88]. Chất nền bên trong nắp sợi đã lành thường bắt đầu dưới dạng khối giàu proteoglycan sớm cùng với collagen loại III/IV, cuối cùng tiến triển thành tổn thương giàu collagen loại I do tổ chức huyết khối (hình 4) [89]. Các vết vỡ đã lành thường biểu hiện nhiều lớp lõi hoại tử xen kẽ bởi mô sợi (còn gọi là mũ chôn), với vị trí vỡ sớm nhất nằm ở lớp nội mạc sâu hơn gợi ý các biến cố huyết khối trước đó, tuần tự dẫn đến tiến triển tổn thương [89].
Một nghiên cứu năm 2019 sử dụng phương pháp chụp cắt lớp mạch lạc quang học hình ảnh nội mạch cho thấy những bệnh nhân mắc ACS tái phát có kiểu hình xơ vữa động mạch khác biệt so với những người mắc chứng đau thắt ngực ổn định mãn tính, bao gồm tỷ lệ mảng bám mạch vành đã lành thấp hơn nhiều. Những phát hiện này cho thấy rằng mặc dù kiểu hình và gánh nặng của bệnh xơ vữa động mạch là những yếu tố quan trọng ảnh hưởng đến bệnh mạch vành cấp tính, các yếu tố khác, bao gồm cả việc lành mảng bám, có thể xác định diễn biến tự nhiên của bệnh nhân đối với việc tái phát các biến cố cấp tính hoặc sự ổn định lâm sàng lâu dài. Do đó, chiến lược chữa lành mảng bám có thể trở thành mục tiêu điều trị mới [90,91]. Người ta đã chứng minh rằng mức độ thu hẹp lòng ống tăng lên khi số lần vỡ tăng lên [88,89].
XÓI MÒN MẢNG BÁM
Xói mòn mảng bám là tổn thương phổ biến thứ hai sau vỡ mảng bám liên quan đến huyết khối cấp tính trong tuần hoàn mạch vành. Xói mòn khác với tổn thương vỡ ở chỗ không có sự phá vỡ mũ xơ (hình 1).
Các đặc điểm tế bào chính của sự xói mòn mảng bám bao gồm sự phong phú của các tế bào cơ trơn trong ma trận giàu proteoglycan và không có nội mô bề mặt mà không có lõi lipid nổi bật [92]. Nhìn chung có rất ít đại thực bào và tế bào lympho T ở gần lòng [1]. Hình thái tổn thương cơ bản cũng khác với vỡ vì nó liên quan đến các tổn thương sớm như dày nội mạc bệnh lý hoặc u xơ vữa động mạch mà không có lõi hoại tử lan rộng, xuất huyết hoặc vôi hóa. Không có thông tin liên lạc giữa lõi hoại tử và lòng. Thường có một vùng giàu tế bào cơ trơn gần lòng mạch ngăn cách huyết khối với lõi hoại tử hoặc bể lipid bên dưới. Sự vắng mặt của nội mạc, thứ phát do mất tế bào nội mô theo chương trình, cho phép máu chảy tiếp xúc với collagen dẫn đến hình thành huyết khối.
Sự tích tụ mạnh mẽ của các phân tử ma trận ngoại bào như hyaluronan và versican (proteoglycan) được quan sát thấy tại các vị trí ăn mòn mảng bám, trong khi có sự tích tụ tương đối ít của proteoglycan và hyaluronan tại các vị trí vỡ mảng bám [92]. Sự xáo trộn dòng chảy có thể thúc đẩy hoạt hóa nội mô với biểu hiện TLR-2, sự tích lũy có chọn lọc của hyaluronan, phối tử của TLR-2 và huy động bạch cầu trung tính, do đó thúc đẩy quá trình khử nội mô và hình thành huyết khối giàu bạch cầu trung tính [93,94]. Thật vậy, các nghiên cứu sau khi chết đã chỉ ra rằng huyết khối mạch vành chồng lên các mảng bị xói mòn chứa mật độ tế bào dương tính với myeloperoxidase cao hơn nhiều so với huyết khối chồng lên các mảng bị vỡ [95]. Nồng độ myeloperoxidase toàn thân cũng tăng ở những bệnh nhân mắc ACS có mảng bám bị xói mòn so với những bệnh nhân bị vỡ, cho thấy rằng sự gia tăng các dấu ấn sinh học viêm chọn lọc có thể phản ánh các biến cố mạch vành cấp tính cụ thể [95,96]. Một nghiên cứu năm 2018 cho thấy sự biểu hiện gia tăng của hyaluronidase-2, chất này biến hyaluronan trọng lượng phân tử cao thành hyaluronan trọng lượng phân tử thấp gây viêm trong tế bào đơn nhân máu ngoại vi của bệnh nhân bị xói mòn mảng bám được đánh giá bằng chụp cắt lớp kết hợp quang học. Sự giải phóng cục bộ hyaluronan trọng lượng phân tử thấp có thể trực tiếp thúc đẩy quá trình trùng hợp fibrin, điều này có thể tạo điều kiện cho sự di chuyển tế bào cơ trơn và phát triển mảng bám, cũng như kích thích TLR-2 và hình thành các tập hợp bạch cầu trung tính-tiểu cầu, thúc đẩy hình thành huyết khối [97,98] .
Các yếu tố nguy cơ xói mòn chưa được hiểu rõ và khác với các yếu tố gây vỡ. Người ta đã suy đoán rằng co thắt mạch vành có thể liên quan đến sinh lý bệnh của xói mòn [99]. Dữ liệu từ các bệnh nhân bị co thắt động mạch vành được chứng minh bằng hình ảnh chụp cắt lớp mạch lạc quang học cho thấy hiện tượng trợt xảy ra ở hơn 1/4 số bệnh nhân với nhiều bệnh nhân trong số này có chứa huyết khối [100].
Các nốt vôi hóa – Các mảng vôi hóa được đặc trưng bởi sự hiện diện của các nốt canxi dày đặc phá vỡ bề mặt lòng ống và nhô vào trong lòng [1,101,102]. Các tổn thương vôi hóa rất khó xác định in vivo do độ phân giải hạn chế của chụp động mạch nhưng đã được xác định ở khoảng 5 đến 8% bệnh nhân mắc ACS bằng chụp cắt lớp kết hợp quang học [103,104]. Những tổn thương này thường phổ biến hơn ở những người lớn tuổi có động mạch vành quanh co và bị vôi hóa nặng và ở động mạch vành phải. Các động mạch bị vôi hóa nặng thường có các mảng vôi hóa lớn với các vùng xơ hóa, viêm và tân mạch xung quanh, có thể được hình thành do sự phân mảnh của lõi canxi hoại tử.
TƯƠNG QUAN LÂM SÀNG
Như đã thảo luận ở trên, vỡ mảng bám và xói mòn mảng bám lần lượt là bệnh lý thứ nhất và thứ hai phổ biến nhất liên quan đến ACS.
Huyết khối trong lòng mạch sau khi máu tiếp xúc với các nốt vôi hóa đã được quan sát thấy. Tuy nhiên, tỷ lệ mắc bệnh ở những nạn nhân đột tử do mạch vành là dưới 5%. Các nghiên cứu hình ảnh đã cho thấy tỷ lệ mắc bệnh cao hơn lên tới 10%, có thể là do bệnh nhân còn sống mắc ACS lớn tuổi hơn nạn nhân tử vong do mạch vành đột ngột [105,106]. Bất kể bệnh lý tiềm ẩn, sự hình thành huyết khối trong lòng mạch là nguyên nhân gây ra ACS.
Các nguyên nhân khác của ACS đã được trình bày ở trên.
Trong trường hợp khi vỡ (hoặc xói mòn) không dẫn đến tắc nghẽn mạch do huyết khối, các triệu chứng có thể không xuất hiện và quá trình lành mảng bám sẽ dẫn đến tiến triển mảng bám và thu hẹp lòng mạch nhiều hơn [88,89].
Các nghiên cứu khám nghiệm tử thi đã chỉ ra rằng khi xác định được huyết khối trong lòng mạch ở bệnh nhân đột tử do tim và nhồi máu cơ tim cấp tính (MI), bệnh lý cơ bản là vỡ 55 đến 75% các trường hợp, xói mòn 25 đến 40% trường hợp và các nốt vôi hóa ít hơn. hơn 5 phần trăm thời gian [1,39,69,76-78,107-109]. Các nghiên cứu in vivo ở những bệnh nhân bị nhồi máu cơ tim cấp ST chênh lên (STEMI) sử dụng phương pháp chụp cắt lớp mạch lạc quang học có độ phân giải cao (OCT) của các mảng thủ phạm đã cho thấy sự phân bố tương tự về hình thái mảng bám [105].
Trong STEMI cấp tính, khoảng 75% trường hợp là do vỡ mảng bám và 25% còn lại là do mảng bám bị xói mòn [76]. Phát hiện này đã được xác nhận bởi các nghiên cứu lâm sàng sử dụng OCT có độ phân giải cao [77].
Trong ACS không ST chênh lên sử dụng OCT, vỡ mảng bám được tìm thấy ở 47% trường hợp [78]. Những phát hiện này đã được xác nhận bằng siêu âm nội mạch [110].
Chủ yếu dựa trên kết quả khám nghiệm tử thi từ các trường hợp tử vong đột ngột do bệnh mạch vành, các vết vỡ đã lành góp phần làm tăng đáng kể gánh nặng mảng bám và thu hẹp lòng mạch. Chúng thường xảy ra khi không có triệu chứng về tim. Những vết vỡ lần đầu dẫn đến tử vong đột ngột do tim chỉ là nguyên nhân gây ra 11% huyết khối cấp tính [89]. Ở những mảng có đường kính hẹp dưới 20%, tỷ lệ vỡ mảng bám đã lành là 16%; ở những tổn thương có mức độ hẹp từ 21 đến 50%, tỷ lệ mắc bệnh là 19%; và ở những mảng có độ thu hẹp lớn hơn 50%, tỷ lệ mắc bệnh là 73% [88].
Giảm cholesterol lipoprotein mật độ thấp, đặc biệt khi điều trị bằng statin, có thể thay đổi đặc điểm của mảng xơ vữa động mạch theo cách làm dày lớp xơ, giảm tích tụ lipid, xua tan tình trạng viêm và thu nhỏ thể tích của lõi lipid. Những thay đổi về hình thái này sẽ dẫn đến sự “ổn định” của các mảng bám, giảm nguy cơ vỡ. Vấn đề này được thảo luận riêng.
Quan sát hình ảnh – Vỡ mảng bám được cho là xuất hiện khi chụp động mạch vành cho thấy các tổn thương phức tạp, bao gồm hẹp với đường viền không đều, gồ ghề và loét có hoặc không có khuyết tật lấp đầy lòng mạch gợi ý huyết khối [111-113]. Sự hiện diện của các tổn thương phức tạp trên chụp động mạch có liên quan đến sự tiến triển nhanh chóng của hẹp động mạch vành [114-116]. Một nghiên cứu OCT ở bệnh nhân mắc ACS cho thấy tỷ lệ vỡ mảng bám là 44%, xói mòn mảng bám là 31% và nốt vôi hóa là 8% [103]. Những phát hiện tương tự cũng được quan sát thấy trong một nghiên cứu khác [105].
Một tỷ lệ đáng kể (40%) bệnh nhân nhồi máu cơ tim cấp tính (MI) có bằng chứng chụp động mạch vành về nhiều tổn thương phức tạp; điều này được gọi là “sự mất ổn định của nhiều mảng bám” [117]. Tình trạng này có liên quan đến kết quả lâm sàng bất lợi. Các nghiên cứu nội soi mạch ủng hộ quan điểm này bằng cách cho thấy sự hiện diện của nhiều mảng màu vàng, tức là các mảng dễ bị tổn thương, khắp cây vành ở bệnh nhân nhồi máu cơ tim cấp tính [118]. Hơn nữa, hình ảnh siêu âm nội mạch ba mạch máu đã chứng minh rằng vỡ mảng bám thủ phạm, vỡ mảng bám từ xa và vỡ nhiều mảng bám đều thường gặp hơn ở những bệnh nhân bị nhồi máu cơ tim cấp so với những bệnh nhân bị đau thắt ngực ổn định [119]. Tuy nhiên, trong các nghiên cứu khám nghiệm tử thi về hình thái học, tỷ lệ xuất hiện nhiều vết vỡ ở một cá nhân là dưới 5% [120]. Những phát hiện tương tự đã được OCT xác nhận cho thấy tổn thương toàn mạch vành ở những bệnh nhân có mảng bám thủ phạm bị vỡ nhưng không xảy ra ở những bệnh nhân có mảng bám thủ phạm không bị nứt.
PHÂN LOẠI BỆNH HỌC CỦA HỘI CHỨNG VÀNH CẤP
Một phân loại bệnh sinh của ACS đã đề xuất phân chia huyết khối động mạch vành do vỡ mảng bám thành các trường hợp có hoặc không có dấu hiệu viêm đồng thời [121]. Sự khác biệt này có thể có ý nghĩa điều trị đáng kể khi các biện pháp can thiệp chống viêm trực tiếp đối với chứng xơ vữa động mạch xuất hiện, đặc biệt là các mảng vỡ mảng bám, có thể được phân biệt bằng chụp cắt lớp mạch lạc quang học. Hiện tại, sự khác biệt này chưa được chứng minh trên lâm sàng [122].
Cơ chế thứ ba là sự xói mòn mảng bám có thể đang gia tăng trong thời kỳ lượng lipid giảm mạnh. Việc xác định bệnh nhân mắc ACS do xói mòn có thể cho phép tiếp cận quản lý ít xâm lấn hơn so với tiêu chuẩn chăm sóc.
Cơ chế thứ tư được thể hiện bằng sự thay đổi chức năng của tuần hoàn mạch vành, bao gồm co thắt thượng tâm mạc và vi mạch, thường xảy ra trong ACS xảy ra mà không có huyết khối hoặc hẹp động mạch vành thượng tâm mạc rõ ràng. Các chiến lược quản lý mới nổi cũng có thể áp dụng có chọn lọc cho loại ACS này. Cách tiếp cận cơ học hơn để phân loại ACS này cung cấp một khuôn khổ cho việc điều chỉnh, phân loại và điều trị trong tương lai cho bệnh nhân theo cách cá nhân hóa và chính xác hơn (hình 4).
TÓM TẮT
●Hầu hết các hội chứng mạch vành cấp tính (ACS) là do mất tính toàn vẹn của lớp vỏ bảo vệ trên mảng xơ vữa động mạch. Việc mất lớp phủ nội mô bảo vệ cho phép máu tiếp xúc với các thành phần có khả năng gây huyết khối cao của collagen và/hoặc lõi hoại tử của mảng bám và tạo điều kiện cho huyết khối ở lòng mạch xảy ra.
●Vỡ mảng bám là nguyên nhân chính gây ra hội chứng mạch vành cấp tính do huyết khối. Nó được đặc trưng bởi các tổn thương biểu hiện các lõi hoại tử tương đối lớn với nắp sợi mỏng bị đứt gãy. Máu ở lòng mạch tiếp xúc trực tiếp với lõi hoại tử và có thể dẫn đến huyết khối.
●Ở một nhóm lớn bệnh nhân, ACS là do xói mòn mảng bám. Sinh bệnh học của sự xói mòn mảng bám khác với vết nứt mảng bám và được đặc trưng bởi sự tích tụ hyaluronan và bạch cầu trung tính, dẫn đến khử nội mô và hình thành huyết khối.
●ACS trong trường hợp không có vết nứt hoặc vỡ mảng xơ vữa động mạch có thể được gây ra bởi bóc tách động mạch vành, co thắt động mạch vành và rối loạn chức năng vi mạch vành. Tỷ lệ mắc các cơ chế mất ổn định mạch vành này thấp hơn so với nứt hoặc xói mòn mảng bám nhưng có thể bị đánh giá thấp.
TÀI LIỆU THAM KHẢO
- Virmani R, Kolodgie FD, Burke AP, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2000; 20:1262.
- Yahagi K, Kolodgie FD, Otsuka F, et al. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol 2016; 13:79.
- Velican D, Velican C. Atherosclerotic involvement of the coronary arteries of adolescents and young adults. Atherosclerosis 1980; 36:449.
- McGill HC Jr, McMahan CA, Zieske AW, et al. Associations of coronary heart disease risk factors with the intermediate lesion of atherosclerosis in youth. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb Vasc Biol 2000; 20:1998.
- Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994; 89:2462.
- Kolodgie FD, Burke AP, Nakazawa G, Virmani R. Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler Thromb Vasc Biol 2007; 27:986.
- Libby P. Inflammation in atherosclerosis. Nature 2002; 420:868.
- Libby P. Changing concepts of atherogenesis. J Intern Med 2000; 247:349.
- Babior BM. Phagocytes and oxidative stress. Am J Med 2000; 109:33.
- Endemann G, Stanton LW, Madden KS, et al. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem 1993; 268:11811.
- Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med 2002; 8:1235.
- Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med 2002; 8:1211.
- Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med 2001; 11:93.
- Pearson AM. Scavenger receptors in innate immunity. Curr Opin Immunol 1996; 8:20.
- Muzio M, Mantovani A. Toll-like receptors (TLRs) signalling and expression pattern. J Endotoxin Res 2001; 7:297.
- Faure E, Thomas L, Xu H, et al. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol 2001; 166:2018.
- Xu XH, Shah PK, Faure E, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001; 104:3103.
- Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010; 11:155.
- Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995; 92:1355.
- Kruth HS. Localization of unesterified cholesterol in human atherosclerotic lesions. Demonstration of filipin-positive, oil-red-O-negative particles. Am J Pathol 1984; 114:201.
- Guyton JR, Klemp KF. Development of the lipid-rich core in human atherosclerosis. Arterioscler Thromb Vasc Biol 1996; 16:4.
- Katz SS, Shipley GG, Small DM. Physical chemistry of the lipids of human atherosclerotic lesions. Demonstration of a lesion intermediate between fatty streaks and advanced plaques. J Clin Invest 1976; 58:200.
- Felton CV, Crook D, Davies MJ, Oliver MF. Relation of plaque lipid composition and morphology to the stability of human aortic plaques. Arterioscler Thromb Vasc Biol 1997; 17:1337.
- Tabas I. Free cholesterol-induced cytotoxicity a possible contributing factor to macrophage foam cell necrosis in advanced atherosclerotic lesions. Trends Cardiovasc Med 1997; 7:256.
- Kockx MM. Apoptosis in the atherosclerotic plaque: quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol 1998; 18:1519.
- Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol 2005; 25:2255.
- Schrijvers DM, De Meyer GR, Kockx MM, et al. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol 2005; 25:1256.
- Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med 2003; 349:2316.
- Sluimer JC, Kolodgie FD, Bijnens AP, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol 2009; 53:1517.
- Boyle JJ, Harrington HA, Piper E, et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol 2009; 174:1097.
- Finn AV, Nakano M, Polavarapu R, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol 2012; 59:166.
- Guo L, Akahori H, Harari E, et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest 2018; 128:1106.
- Otsuka F, Sakakura K, Yahagi K, et al. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol 2014; 34:724.
- Glagov S, Weisenberg E, Zarins CK, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 1987; 316:1371.
- Clarkson TB, Prichard RW, Morgan TM, et al. Remodeling of coronary arteries in human and nonhuman primates. JAMA 1994; 271:289.
- Burke AP, Kolodgie FD, Farb A, et al. Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation 2002; 105:297.
- Varnava AM, Mills PG, Davies MJ. Relationship between coronary artery remodeling and plaque vulnerability. Circulation 2002; 105:939.
- Kubo T, Maehara A, Mintz GS, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol 2010; 55:1590.
- Burke AP, Farb A, Malcom GT, et al. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med 1997; 336:1276.
- Davies MJ, Thomas AC. Plaque fissuring–the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J 1985; 53:363.
- Kolodgie FD, Burke AP, Farb A, et al. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol 2001; 16:285.
- Kolodgie FD, Virmani R, Burke AP, et al. Pathologic assessment of the vulnerable human coronary plaque. Heart 2004; 90:1385.
- Siegel RJ, Swan K, Edwalds G, Fishbein MC. Limitations of postmortem assessment of human coronary artery size and luminal narrowing: differential effects of tissue fixation and processing on vessels with different degrees of atherosclerosis. J Am Coll Cardiol 1985; 5:342.
- Tanaka A, Imanishi T, Kitabata H, et al. Morphology of exertion-triggered plaque rupture in patients with acute coronary syndrome: an optical coherence tomography study. Circulation 2008; 118:2368.
- Yonetsu T, Kakuta T, Lee T, et al. In vivo critical fibrous cap thickness for rupture-prone coronary plaques assessed by optical coherence tomography. Eur Heart J 2011; 32:1251.
- Burke AP, Farb A, Malcom GT, et al. Plaque rupture and sudden death related to exertion in men with coronary artery disease. JAMA 1999; 281:921.
- Nemerson Y. A simple experiment and a weakening paradigm: the contribution of blood to propensity for thrombus formation. Arterioscler Thromb Vasc Biol 2002; 22:1369.
- Flego D, Liuzzo G, Weyand CM, Crea F. Adaptive Immunity Dysregulation in Acute Coronary Syndromes: From Cellular and Molecular Basis to Clinical Implications. J Am Coll Cardiol 2016; 68:2107.
- Angelini G, Flego D, Vinci R, et al. Matrix metalloproteinase-9 might affect adaptive immunity in non-ST segment elevation acute coronary syndromes by increasing CD31 cleavage on CD4+ T-cells. Eur Heart J 2018; 39:1089.
- Libby P. Molecular bases of the acute coronary syndromes. Circulation 1995; 91:2844.
- Lutgens E, van Suylen RJ, Faber BC, et al. Atherosclerotic plaque rupture: local or systemic process? Arterioscler Thromb Vasc Biol 2003; 23:2123.
- Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21:1876.
- Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res 2002; 91:281.
- Libby P. Coronary artery injury and the biology of atherosclerosis: inflammation, thrombosis, and stabilization. Am J Cardiol 2000; 86:3J.
- Dollery CM, Owen CA, Sukhova GK, et al. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation 2003; 107:2829.
- Herman MP, Sukhova GK, Libby P, et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation 2001; 104:1899.
- Sukhova GK, Schönbeck U, Rabkin E, et al. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 1999; 99:2503.
- Sukhova GK, Shi GP, Simon DI, et al. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest 1998; 102:576.
- Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res 2002; 90:251.
- Kolodgie FD, Narula J, Guillo P, Virmani R. Apoptosis in human atherosclerotic plaques. Apoptosis 1999; 4:5.
- Geng YJ, Libby P. Progression of atheroma: a struggle between death and procreation. Arterioscler Thromb Vasc Biol 2002; 22:1370.
- Geng YJ, Henderson LE, Levesque EB, et al. Fas is expressed in human atherosclerotic intima and promotes apoptosis of cytokine-primed human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1997; 17:2200.
- Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest 1995; 95:2266.
- Kolodgie FD, Narula J, Burke AP, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol 2000; 157:1259.
- Farb A, Burke AP, Tang AL, et al. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation 1996; 93:1354.
- Janoudi A, Shamoun FE, Kalavakunta JK, Abela GS. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur Heart J 2016; 37:1959.
- Buffon A, Biasucci LM, Liuzzo G, et al. Widespread coronary inflammation in unstable angina. N Engl J Med 2002; 347:5.
- Lombardo A, Biasucci LM, Lanza GA, et al. Inflammation as a possible link between coronary and carotid plaque instability. Circulation 2004; 109:3158.
- Burke AP, Farb A, Malcom GT, et al. Effect of risk factors on the mechanism of acute thrombosis and sudden coronary death in women. Circulation 1998; 97:2110.
- Loree HM, Kamm RD, Stringfellow RG, Lee RT. Effects of fibrous cap thickness on peak circumferential stress in model atherosclerotic vessels. Circ Res 1992; 71:850.
- Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999; 282:2035.
- Gijsen FJ, Wentzel JJ, Thury A, et al. Strain distribution over plaques in human coronary arteries relates to shear stress. Am J Physiol Heart Circ Physiol 2008; 295:H1608.
- Slager CJ, Wentzel JJ, Gijsen FJ, et al. The role of shear stress in the generation of rupture-prone vulnerable plaques. Nat Clin Pract Cardiovasc Med 2005; 2:401.
- Vengrenyuk Y, Carlier S, Xanthos S, et al. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci U S A 2006; 103:14678.
- Kelly-Arnold A, Maldonado N, Laudier D, et al. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci U S A 2013; 110:10741.
- Arbustini E, Dal Bello B, Morbini P, et al. Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction. Heart 1999; 82:269.
- Kubo T, Imanishi T, Takarada S, et al. Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J Am Coll Cardiol 2007; 50:933.
- Ino Y, Kubo T, Tanaka A, et al. Difference of culprit lesion morphologies between ST-segment elevation myocardial infarction and non-ST-segment elevation acute coronary syndrome: an optical coherence tomography study. JACC Cardiovasc Interv 2011; 4:76.
- Achenbach S, Ulzheimer S, Baum U, et al. Noninvasive coronary angiography by retrospectively ECG-gated multislice spiral CT. Circulation 2000; 102:2823.
- Hoffmann MH, Shi H, Schmitz BL, et al. Noninvasive coronary angiography with multislice computed tomography. JAMA 2005; 293:2471.
- Schroeder S, Kopp AF, Baumbach A, et al. Noninvasive detection and evaluation of atherosclerotic coronary plaques with multislice computed tomography. J Am Coll Cardiol 2001; 37:1430.
- Leber AW, Knez A, Becker A, et al. Accuracy of multidetector spiral computed tomography in identifying and differentiating the composition of coronary atherosclerotic plaques: a comparative study with intracoronary ultrasound. J Am Coll Cardiol 2004; 43:1241.
- Hoffmann U, Moselewski F, Nieman K, et al. Noninvasive assessment of plaque morphology and composition in culprit and stable lesions in acute coronary syndrome and stable lesions in stable angina by multidetector computed tomography. J Am Coll Cardiol 2006; 47:1655.
- Motoyama S, Sarai M, Harigaya H, et al. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. J Am Coll Cardiol 2009; 54:49.
- Motoyama S, Ito H, Sarai M, et al. Plaque Characterization by Coronary Computed Tomography Angiography and the Likelihood of Acute Coronary Events in Mid-Term Follow-Up. J Am Coll Cardiol 2015; 66:337.
- Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011; 364:226.
- Erlinge D, Maehara A, Ben-Yehuda O, et al. Identification of vulnerable plaques and patients by intracoronary near-infrared spectroscopy and ultrasound (PROSPECT II): a prospective natural history study. Lancet 2021; 397:985.
- Mann J, Davies MJ. Mechanisms of progression in native coronary artery disease: role of healed plaque disruption. Heart 1999; 82:265.
- Burke AP, Kolodgie FD, Farb A, et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation 2001; 103:934.
- Vergallo R, Porto I, D’Amario D, et al. Coronary Atherosclerotic Phenotype and Plaque Healing in Patients With Recurrent Acute Coronary Syndromes Compared With Patients With Long-term Clinical Stability: An In Vivo Optical Coherence Tomography Study. JAMA Cardiol 2019; 4:321.
- Vergallo R, Crea F. Atherosclerotic Plaque Healing. N Engl J Med 2020; 383:846.
- Kolodgie FD, Burke AP, Farb A, et al. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler Thromb Vasc Biol 2002; 22:1642.
- Franck G, Mawson T, Sausen G, et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ Res 2017; 121:31.
- Quillard T, Araújo HA, Franck G, et al. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J 2015; 36:1394.
- Ferrante G, Nakano M, Prati F, et al. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 2010; 122:2505.
- Kramer MC, Rittersma SZ, de Winter RJ, et al. Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J Am Coll Cardiol 2010; 55:122.
- Pedicino D, Vinci R, Giglio AF, et al. Alterations of Hyaluronan Metabolism in Acute Coronary Syndrome: Implications for Plaque Erosion. J Am Coll Cardiol 2018; 72:1490.
- Vinci R, Pedicino D, D’Aiello A, et al. Platelet hyaluronidase 2 enrichment in acute coronary syndromes: a conceivable role in monocyte-platelet aggregate formation. J Enzyme Inhib Med Chem 2021; 36:785.
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol 2006; 47:C13.
- Shin ES, Ann SH, Singh GB, et al. OCT-Defined Morphological Characteristics of Coronary Artery Spasm Sites in Vasospastic Angina. JACC Cardiovasc Imaging 2015; 8:1059.
- Torii S, Sato Y, Otsuka F, et al. Eruptive Calcified Nodules as a Potential Mechanism of Acute Coronary Thrombosis and Sudden Death. J Am Coll Cardiol 2021; 77:1599.
- Arbustini E, Vengrenyuk Y, Narula J. On the Shades of Coronary Calcium and Plaque Instability. J Am Coll Cardiol 2021; 77:1612.
- Jia H, Abtahian F, Aguirre AD, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol 2013; 62:1748.
- Sugane H, Kataoka Y, Otsuka F, et al. Cardiac outcomes in patients with acute coronary syndrome attributable to calcified nodule. Atherosclerosis 2021; 318:70.
- Higuma T, Soeda T, Abe N, et al. A Combined Optical Coherence Tomography and Intravascular Ultrasound Study on Plaque Rupture, Plaque Erosion, and Calcified Nodule in Patients With ST-Segment Elevation Myocardial Infarction: Incidence, Morphologic Characteristics, and Outcomes After Percutaneous Coronary Intervention. JACC Cardiovasc Interv 2015; 8:1166.
- Chin CY, Matsumura M, Maehara A, et al. Coronary Plaque Characteristics in Hemodialysis-Dependent Patients as Assessed by Optical Coherence Tomography. Am J Cardiol 2017; 119:1313.
- Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med 1984; 310:1137.
- el Fawal MA, Berg GA, Wheatley DJ, Harland WA. Sudden coronary death in Glasgow: nature and frequency of acute coronary lesions. Br Heart J 1987; 57:329.
- Davies MJ, Bland JM, Hangartner JR, et al. Factors influencing the presence or absence of acute coronary artery thrombi in sudden ischaemic death. Eur Heart J 1989; 10:203.
- Hong YJ, Jeong MH, Choi YH, et al. Differences in intravascular ultrasound findings in culprit lesions in infarct-related arteries between ST segment elevation myocardial infarction and non-ST segment elevation myocardial infarction. J Cardiol 2010; 56:15.
- Levin DC, Fallon JT. Significance of the angiographic morphology of localized coronary stenoses: histopathologic correlations. Circulation 1982; 66:316.
- Ambrose JA, Winters SL, Stern A, et al. Angiographic morphology and the pathogenesis of unstable angina pectoris. J Am Coll Cardiol 1985; 5:609.
- Ambrose JA, Winters SL, Arora RR, et al. Angiographic evolution of coronary artery morphology in unstable angina. J Am Coll Cardiol 1986; 7:472.
- Kaski JC, Chester MR, Chen L, Katritsis D. Rapid angiographic progression of coronary artery disease in patients with angina pectoris. The role of complex stenosis morphology. Circulation 1995; 92:2058.
- Chen L, Chester MR, Redwood S, et al. Angiographic stenosis progression and coronary events in patients with ‘stabilized’ unstable angina. Circulation 1995; 91:2319.
- Chester MR, Chen L, Kaski JC. The natural history of unheralded complex coronary plaques. J Am Coll Cardiol 1996; 28:604.
- Goldstein JA, Demetriou D, Grines CL, et al. Multiple complex coronary plaques in patients with acute myocardial infarction. N Engl J Med 2000; 343:915.
- Asakura M, Ueda Y, Yamaguchi O, et al. Extensive development of vulnerable plaques as a pan-coronary process in patients with myocardial infarction: an angioscopic study. J Am Coll Cardiol 2001; 37:1284.
- Hong MK, Mintz GS, Lee CW, et al. Comparison of coronary plaque rupture between stable angina and acute myocardial infarction: a three-vessel intravascular ultrasound study in 235 patients. Circulation 2004; 110:928.
- Burke A, Virmani R. Significance of multiple coronary artery thrombi. A consequence of diffuse atherosclerotic disease? Ital Heart J 2000; 1:832.
- Crea F, Libby P. Acute Coronary Syndromes: The Way Forward From Mechanisms to Precision Treatment. Circulation 2017; 136:1155.
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017; 377:1119.
BÌNH LUẬN